 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5dlx: FIRST DOMAIN OF HUMAN BROMODOMAIN BRD4 IN COMPLEX WITH INHIBITOR ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dlx | ||||||
|---|---|---|---|---|---|---|---|
| Title | FIRST DOMAIN OF HUMAN BROMODOMAIN BRD4 IN COMPLEX WITH INHIBITOR N-{3-[4-(3-chlorophenyl)piperazin-1-yl]propyl}-1-{3-methyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidine-4-carboxamide | ||||||
 Components Components | Bromodomain-containing protein 4 | ||||||
 Keywords Keywords | TRANSCRIPTION / INHIBITOR / HISTONE / EPIGENETIC READER / BROMODOMAIN / BRD4 / Brd4_BD1 | ||||||
| Function / homology |  Function and homology information Function and homology informationRNA polymerase II C-terminal domain binding / P-TEFb complex binding / negative regulation of DNA damage checkpoint / histone H4 reader activity / host-mediated suppression of viral transcription / positive regulation of G2/M transition of mitotic cell cycle / positive regulation of T-helper 17 cell lineage commitment / : / RNA polymerase II CTD heptapeptide repeat kinase activity / condensed nuclear chromosome ...RNA polymerase II C-terminal domain binding / P-TEFb complex binding / negative regulation of DNA damage checkpoint / histone H4 reader activity / host-mediated suppression of viral transcription / positive regulation of G2/M transition of mitotic cell cycle / positive regulation of T-helper 17 cell lineage commitment / : / RNA polymerase II CTD heptapeptide repeat kinase activity / condensed nuclear chromosome / transcription coregulator activity / positive regulation of transcription elongation by RNA polymerase II / p53 binding / chromosome / regulation of inflammatory response / histone binding / Potential therapeutics for SARS / transcription coactivator activity / positive regulation of canonical NF-kappaB signal transduction / transcription cis-regulatory region binding / chromatin remodeling / protein serine/threonine kinase activity / DNA damage response / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / positive regulation of DNA-templated transcription / enzyme binding / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Raux, B. / Rebuffet, E. / Betzi, S. / Morelli, X. | ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2016 Title: Protein-Protein Interaction Inhibition (2P2I)-Oriented Chemical Library Accelerates Hit Discovery. Authors: Milhas, S. / Raux, B. / Betzi, S. / Derviaux, C. / Roche, P. / Restouin, A. / Basse, M.J. / Rebuffet, E. / Lugari, A. / Badol, M. / Kashyap, R. / Lissitzky, J.C. / Eydoux, C. / Hamon, V. / ...Authors: Milhas, S. / Raux, B. / Betzi, S. / Derviaux, C. / Roche, P. / Restouin, A. / Basse, M.J. / Rebuffet, E. / Lugari, A. / Badol, M. / Kashyap, R. / Lissitzky, J.C. / Eydoux, C. / Hamon, V. / Gourdel, M.E. / Combes, S. / Zimmermann, P. / Aurrand-Lions, M. / Roux, T. / Rogers, C. / Muller, S. / Knapp, S. / Trinquet, E. / Collette, Y. / Guillemot, J.C. / Morelli, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dlx.cif.gz | 72.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dlx.ent.gz | 52.3 KB | Display | PDB format |
| PDBx/mmJSON format | 5dlx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5dlx_validation.pdf.gz | 649 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5dlx_full_validation.pdf.gz | 651 KB | Display | |
| Data in XML | 5dlx_validation.xml.gz | 7.9 KB | Display | |
| Data in CIF | 5dlx_validation.cif.gz | 9.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dl/5dlxftp://data.pdbj.org/pub/pdb/validation_reports/dl/5dlx | HTTPS FTP |
-Related structure data
| Related structure data |  5dlzC  2ossS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 15099.380 Da / Num. of mol.: 1 / Fragment: N-TERMINAL BROMODOMAIN, RESIDUES 42-168 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BRD4, HUNK1 / Plasmid: PLASMID / Details (production host): pDEST17 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-5D2 / 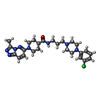  Mass: 497.036 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H33ClN8O Mass: 497.036 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H33ClN8O |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.96 Å3/Da / Density % sol: 37.31 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: Ratio 1:1 (protein:precipitant). 12 mg/mL Brd4_BD1 protein. Protein buffer 10 mM HEPES (pH 7.5) 150 mM NaCl. Precipitant agent 22 % (w/v) PEG 3350, 10% (w/v) ethylene glycol 30 % (w/v), 0.3 ...Details: Ratio 1:1 (protein:precipitant). 12 mg/mL Brd4_BD1 protein. Protein buffer 10 mM HEPES (pH 7.5) 150 mM NaCl. Precipitant agent 22 % (w/v) PEG 3350, 10% (w/v) ethylene glycol 30 % (w/v), 0.3 M NaFormate. 1.0 mM inhibitor (in DMSO) final concentration. Cryoprotection with 10 % (w/v) glycerol PH range: 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.9677 Å / Beamline: ID23-2 / Wavelength: 0.9677 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Apr 15, 2015 |
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9677 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→50 Å / Num. obs: 9664 / % possible obs: 99.3 % / Redundancy: 4.83 % / Rsym value: 0.042 / Net I/σ(I): 20.09 |
| Reflection shell | Resolution: 1.9→2.02 Å / Redundancy: 4.59 % / Mean I/σ(I) obs: 3.73 / Rsym value: 0.379 / % possible all: 97.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2OSS Resolution: 1.9→37.23 Å / Cor.coef. Fo:Fc: 0.984 / Cor.coef. Fo:Fc free: 0.963 / SU B: 9.763 / SU ML: 0.118 / Cross valid method: THROUGHOUT / ESU R Free: 0.14 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.711 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→37.23 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|