 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dbq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of insect thioredoxin at 1.95 Angstroms | |||||||||
 Components Components | Thioredoxin | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / Thioredoxin / Disulfide bridge | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Anticarsia gemmatalis (velvetbean caterpillar) Anticarsia gemmatalis (velvetbean caterpillar) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | |||||||||
 Authors Authors | Klinke, S. / Tejedor, M.D. / Cerutti, M.L. / Giacometti, R. / Otero, L.H. / Goldbaum, F.A. / Zavala, J.A. / Wolosiuk, R.A. / Pagano, E.A. | |||||||||
| Funding support |  Argentina, 2items Argentina, 2items
| |||||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of insect thioredoxin at 1.95 Angstroms Authors: Klinke, S. / Tejedor, M.D. / Cerutti, M.L. / Giacometti, R. / Otero, L.H. / Goldbaum, F.A. / Zavala, J.A. / Wolosiuk, R.A. / Pagano, E.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dbq.cif.gz | 58.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dbq.ent.gz | 41.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5dbq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5dbq_validation.pdf.gz | 426.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5dbq_full_validation.pdf.gz | 427.9 KB | Display | |
| Data in XML | 5dbq_validation.xml.gz | 11.7 KB | Display | |
| Data in CIF | 5dbq_validation.cif.gz | 16.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/db/5dbqftp://data.pdbj.org/pub/pdb/validation_reports/db/5dbq | HTTPS FTP |
-Related structure data
| Related structure data |  1xwbS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 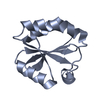
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 12138.050 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Anticarsia gemmatalis (velvetbean caterpillar)Plasmid: pET 28a(+) / Production host:  #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 188 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 188 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | Cloning artifact GSH at the N-terminus after Thrombin cleavage. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.02 Å3/Da / Density % sol: 39.1 % / Description: Small Prisms |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 27% PEG 4000, 0.2 M sodium acetate, 0.1 M Tris pH 8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SEALED TUBE / Type: BRUKER D8 QUEST / Wavelength: 1.5418 Å |
| Detector | Type: BRUKER PHOTON 100 / Detector: PIXEL / Date: Jul 2, 2015 Details: Diffractometer: Bruker D8 QUEST, Source: IMUS Copper Microfocus, Detector: PHOTON 100 |
| Radiation | Monochromator: Helios MX multilayer optics / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→50 Å / Num. obs: 14272 / % possible obs: 98.4 % / Redundancy: 5.2 % / Biso Wilson estimate: 24.01 Å2 / Rmerge(I) obs: 0.127 / Rsym value: 0.127 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 1.95→2.07 Å / Redundancy: 5.1 % / Rmerge(I) obs: 0.735 / Mean I/σ(I) obs: 2 / % possible all: 98.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB 1XWB Resolution: 1.95→31.33 Å / Cor.coef. Fo:Fc: 0.9258 / Cor.coef. Fo:Fc free: 0.9094 / SU R Cruickshank DPI: 0.211 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.233 / SU Rfree Blow DPI: 0.182 / SU Rfree Cruickshank DPI: 0.175
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.41 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.283 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.95→31.33 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.95→2.11 Å / Total num. of bins used: 7
|
