 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5csm | ||||||
|---|---|---|---|---|---|---|---|
| Title | YEAST CHORISMATE MUTASE, T226S MUTANT, COMPLEX WITH TRP | ||||||
 Components Components | CHORISMATE MUTASE | ||||||
 Keywords Keywords | COMPLEX (ISOMERASE/PEPTIDE) / CHORISMATE PYRUVATEMUTASE / ALLOSTERIC PROTEIN / COMPLEX (ISOMERASE-PEPTIDE) / TRANSITION STATE ANALOG / COMPLEX (ISOMERASE-PEPTIDE) complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtryptophan binding / L-tyrosine binding / L-tyrosine biosynthetic process / chorismate metabolic process / chorismate mutase / chorismate mutase activity / L-phenylalanine biosynthetic process / aromatic amino acid biosynthetic process / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Straeter, N. / Schnappauf, G. / Braus, G. / Lipscomb, W.N. | ||||||
 Citation Citation | Journal: Structure / Year: 1997 Title: Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Authors: Strater, N. / Schnappauf, G. / Braus, G. / Lipscomb, W.N. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1996Title: Crystal Structure of the T State of Allosteric Yeast Chorismate Mutase and Comparison with the R State Authors: Strater, N. / Hakansson, K. / Schnappauf, G. / Braus, G. / Lipscomb, W.N. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1995Title: Location of the Active Site of Allosteric Chorismate Mutase from Saccharomyces Cerevisiae, and Comments on the Catalytic and Regulatory Mechanisms Authors: Xue, Y. / Lipscomb, W.N. #3: Journal: Proc.Natl.Acad.Sci.USA / Year: 1994Title: The Crystal Structure of Allosteric Chorismate Mutase at 2.2-A Resolution Authors: Xue, Y. / Lipscomb, W.N. / Graf, R. / Schnappauf, G. / Braus, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5csm.cif.gz | 64.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5csm.ent.gz | 47.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5csm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cs/5csmftp://data.pdbj.org/pub/pdb/validation_reports/cs/5csm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3csmC  4csmC  1csmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29944.457 Da / Num. of mol.: 1 / Mutation: T226S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: RH1242 / Plasmid: PME605 / Production host: |
|---|---|
| #2: Chemical | ChemComp-TRP / 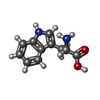  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12N2O2 / Details: TRYPTOPHAN BOUND TO A REGULATORY SITE Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12N2O2 / Details: TRYPTOPHAN BOUND TO A REGULATORY SITE |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 49.25 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 5 Details: HANGING DROP, 19 % PEG 3350, 3 MM DTT, 0.16 M SODIUM ACETATE PH 5.0, 16 MM TRYPTOPHAN, 10 MG/ML PROTEIN, vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ELLIOTT GX-13 / Wavelength: 1.5418 |
| Detector | Type: SIEMENS-NICOLET X100 / Detector: AREA DETECTOR / Date: Jun 27, 1996 / Details: SUPPER DOUBLE-MIRROR, NI-COATED |
| Radiation | Monochromator: DOUBLE CRYSTAL SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. obs: 18951 / % possible obs: 96.6 % / Observed criterion σ(I): 0 / Redundancy: 2.2 % / Rmerge(I) obs: 0.055 |
| Reflection shell | Resolution: 2→2.1 Å / Rmerge(I) obs: 0.196 / % possible all: 88.8 |
| Reflection | *PLUS Num. measured all: 40918 |
| Reflection shell | *PLUS % possible obs: 88.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1CSM Resolution: 2→7 Å / σ(F): 2 Details: RESIDUES THR 218 - GLU 223 ARE NOT VISIBLE IN THE ELECTRON DENSITY MAPS (LOOP REGION). RESIDUES THR 218 - GLU 223 ARE NOT VISIBLE IN THE ELECTRON DENSITY MAPS (LOOP REGION).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.09 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.1 Å / Rfactor obs: 0.255 |
