Mass: 18.015 Da / Num. of mol.: 46 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION
-
Sample preparation
Crystal
Density Matthews: 2.06 Å3/Da / Density % sol: 40.19 % / Description: Crystals were grown as long rods
Crystal grow
Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6 Details: Activated hepsin was incubated with 10 mM Benzamidine at 4 oC for 2 hours and concentrated to7 mg/ml using 10 KDa cut off centricon [Millipore]. 1microlit of protein was mixed with 1 ...Details: Activated hepsin was incubated with 10 mM Benzamidine at 4 oC for 2 hours and concentrated to7 mg/ml using 10 KDa cut off centricon [Millipore]. 1microlit of protein was mixed with 1 microlit of reservoir buffer containing 100 mM sodium Cacodylate (6.0), 22% PEG 3350, and 200 mM ammonium fluoride. Plate clusters with a dimension of 50 micron m x 100 micron m were observed after 2 days of crystallization set up. PH range: 5.8 - 6.3
Resolution: 2.5→58.63 Å / Cor.coef. Fo:Fc: 0.935 / Cor.coef. Fo:Fc free: 0.859 / SU B: 10.623 / SU ML: 0.246 / Cross valid method: THROUGHOUT / ESU R Free: 0.399 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: THE REASON FOR POOR ELECTRON DENSITY IS THAT THE REGION BETWEEN A LEU 158 AND RESIDUE A ILE 163 HIGHLY DISORDERED AND HENCE THE ATOMS POSITIONS ARE NOT STABILIZED IN THE CRYSTAL. HYDROGENS ...Details: THE REASON FOR POOR ELECTRON DENSITY IS THAT THE REGION BETWEEN A LEU 158 AND RESIDUE A ILE 163 HIGHLY DISORDERED AND HENCE THE ATOMS POSITIONS ARE NOT STABILIZED IN THE CRYSTAL. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2815
485
4.8 %
RANDOM
Rwork
0.18714
-
-
-
obs
0.19165
9554
86.66 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj Assembly
Assembly

 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P05981, hepsin
SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P05981, hepsin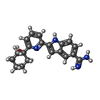
 Mass: 334.415 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22N4O
Mass: 334.415 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22N4O Mass: 18.015 Da / Num. of mol.: 46 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 46 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing