Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
Sequence details
1. SEQUENCE CONFLICTS D270G, A337T AND I479V ARE BASED ON BAB43823.1 ACCORDING TO DATABASE Q9C0L9 ...1. SEQUENCE CONFLICTS D270G, A337T AND I479V ARE BASED ON BAB43823.1 ACCORDING TO DATABASE Q9C0L9 (SEY1_CANAL). 2. THE CODON CUG (MRNA CUG CORRESPONDS TO CTG IN DNA) WILL ENCODE SER IN CANDIDA ALBICANS BUT LEU IN ESCHERICHIA COLI. SO 89S,221S,665S IN CANDIDA ALBICANS SEY1P(BAB43817) WILL BE TRANSLATED INTO LEU IN ESCHERICHIA COLI BL21.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.77 Å3/Da / Density % sol: 55.47 %
Crystal grow
Temperature: 289 K / Method: evaporation Details: 160 mM Lithium sulfate monohydrate, 80 mM Tris pH 8.5, 20% w/v Polyethylene glycol 3,350, 40 mM Ammonium acetate, 20 mM Sodium citrate tribasic dihydrate pH 5.6, 6% w/v Polyethylene glycol ...Details: 160 mM Lithium sulfate monohydrate, 80 mM Tris pH 8.5, 20% w/v Polyethylene glycol 3,350, 40 mM Ammonium acetate, 20 mM Sodium citrate tribasic dihydrate pH 5.6, 6% w/v Polyethylene glycol 4,000, 20 mM spermidine
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1 Å / Relative weight: 1
Reflection
Resolution: 2.8→50 Å / Num. obs: 21774 / % possible obs: 99 % / Redundancy: 10.28 % / Net I/σ(I): 43.9
Reflection shell
Resolution: 2.8→2.85 Å
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0049
refinement
HKL-2000
dataprocessing
PHENIX
modelbuilding
Refinement
Resolution: 2.8→48.76 Å / Cor.coef. Fo:Fc: 0.911 / Cor.coef. Fo:Fc free: 0.902 / SU B: 30.112 / SU ML: 0.316 / Cross valid method: THROUGHOUT / ESU R Free: 0.41 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.27901
1121
5.1 %
RANDOM
Rwork
0.25277
-
-
-
obs
0.2541
20650
98.94 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Candida albicans (yeast)
Candida albicans (yeast) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors China, 3items
China, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj




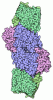

 Assembly
Assembly



 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg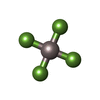
 Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4
Mass: 102.975 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: AlF4
 Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM
Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing