 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4zw8: X-ray crystal structure of PfA-M1 in complex with hydroxamic acid... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zw8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray crystal structure of PfA-M1 in complex with hydroxamic acid-based inhibitor 9r | ||||||
 Components Components | M1 family aminopeptidase | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / M1 ALANYL-AMINOPEPTIDASE / PROTEASE / INHIBITOR / HYDROXAMIC ACID / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-containing vacuole membrane / vacuolar lumen / food vacuole / Hydrolases; Acting on peptide bonds (peptidases); Aminopeptidases / dipeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / proteolysis / zinc ion binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Drinkwater, N. / McGowan, S. | ||||||
| Funding support |  Australia, 1items Australia, 1items
| ||||||
 Citation Citation | Journal: Eur.J.Med.Chem. / Year: 2016 Title: Potent dual inhibitors of Plasmodium falciparum M1 and M17 aminopeptidases through optimization of S1 pocket interactions. Authors: Drinkwater, N. / Vinh, N.B. / Mistry, S.N. / Bamert, R.S. / Ruggeri, C. / Holleran, J.P. / Loganathan, S. / Paiardini, A. / Charman, S.A. / Powell, A.K. / Avery, V.M. / McGowan, S. / Scammells, P.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zw8.cif.gz | 420.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zw8.ent.gz | 337.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4zw8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zw/4zw8ftp://data.pdbj.org/pub/pdb/validation_reports/zw/4zw8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4zw3C  4zw5C  4zw6C  4zw7C  4zx3C  4zx4C  4zx5C  4zx6C  4zx8C  4zx9C  4zy0C  4zy1C  4zy2C  4zyqC  3ebgS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 103760.719 Da / Num. of mol.: 1 / Fragment: UNP residues 195-1084 / Mutation: N213Q, N223Q, H378P, N501Q, N795Q, N1069Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: isolate FcB1 / Columbia / Plasmid: PTRCHIS-2B / Production host:  References: UniProt: O96935, Hydrolases; Acting on peptide bonds (peptidases); Aminopeptidases |
|---|
-Non-polymers , 5 types, 938 molecules 



| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||
|---|---|---|---|---|---|
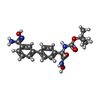
| #3: Chemical | ChemComp-4T2 /  Mass: 400.428 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H24N4O5 Mass: 400.428 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H24N4O5 | ||||
| #4: Chemical | ChemComp-DMS /  Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#5: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 930 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.35 Å3/Da / Density % sol: 47.75 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 22% (v/v) PEG 8000, 10% (v/v) glycerol, 0.1 M Tris pH 8.5, 0.2 M MgCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron / Beamline: MX1 / Wavelength: 0.9537 Å | |||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Aug 14, 2014 | |||||||||||||||||||||||||||
| Radiation | Monochromator: DOUBLE CRYSTAL SILICON 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9537 Å / Relative weight: 1 | |||||||||||||||||||||||||||
| Reflection | Resolution: 2→35.69 Å / Num. obs: 66805 / % possible obs: 99.9 % / Redundancy: 6.7 % / Biso Wilson estimate: 23.86 Å2 / CC1/2: 0.994 / Rmerge(I) obs: 0.158 / Rpim(I) all: 0.065 / Net I/σ(I): 8.5 / Num. measured all: 447176 / Scaling rejects: 41 | |||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3EBG Resolution: 2→35.688 Å / FOM work R set: 0.8604 / SU ML: 0.22 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 20.97 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 102.73 Å2 / Biso mean: 27.11 Å2 / Biso min: 7.34 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→35.688 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 24
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 93.2173 Å / Origin y: 113.0877 Å / Origin z: 128.5903 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
