PROTEIN BINDING / Bacterial chemotaxis / Aspartate receptor / Aspartate-binding protein
Function / homology
Function and homology information
detection of chemical stimulus / methyl accepting chemotaxis protein complex / protein histidine kinase binding / cell tip / cellular response to amino acid stimulus / protein homooligomerization / chemotaxis / transmembrane signaling receptor activity / intracellular signal transduction / protein homodimerization activity ...detection of chemical stimulus / methyl accepting chemotaxis protein complex / protein histidine kinase binding / cell tip / cellular response to amino acid stimulus / protein homooligomerization / chemotaxis / transmembrane signaling receptor activity / intracellular signal transduction / protein homodimerization activity / identical protein binding / plasma membrane Similarity search - Function
Aspartate receptor, ligand-binding domain / Methyl-accepting chemotaxis protein, four helix bundle domain superfamily / Homologues of the ligand binding domain of Tar / Chemotaxis methyl-accepting receptor, methyl-accepting site / Bacterial chemotaxis sensory transducers signature. / Chemotaxis methyl-accepting receptor Tar-related, ligand-binding / Tar ligand binding domain homologue / : / Chemotaxis methyl-accepting receptor / Methyl-accepting chemotaxis protein (MCP) signalling domain ...Aspartate receptor, ligand-binding domain / Methyl-accepting chemotaxis protein, four helix bundle domain superfamily / Homologues of the ligand binding domain of Tar / Chemotaxis methyl-accepting receptor, methyl-accepting site / Bacterial chemotaxis sensory transducers signature. / Chemotaxis methyl-accepting receptor Tar-related, ligand-binding / Tar ligand binding domain homologue / : / Chemotaxis methyl-accepting receptor / Methyl-accepting chemotaxis protein (MCP) signalling domain / Methyl-accepting chemotaxis protein (MCP) signalling domain / Bacterial chemotaxis sensory transducers domain profile. / Methyl-accepting chemotaxis-like domains (chemotaxis sensory transducer). / HAMP domain / HAMP (Histidine kinases, Adenylyl cyclases, Methyl binding proteins, Phosphatases) domain / HAMP domain profile. / HAMP domain / Four Helix Bundle (Hemerythrin (Met), subunit A) / Up-down Bundle / Mainly Alpha Similarity search - Domain/homology
Resolution: 1.57→42 Å / Cor.coef. Fo:Fc: 0.964 / Cor.coef. Fo:Fc free: 0.951 / SU B: 1.236 / SU ML: 0.045 / Cross valid method: THROUGHOUT / ESU R: 0.073 / ESU R Free: 0.076 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.19849
2590
4.9 %
RANDOM
Rwork
0.16639
-
-
-
obs
0.16793
50227
99.5 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj


 Assembly
Assembly
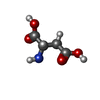
 Type: L-peptide linking / Mass: 133.103 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO4
Type: L-peptide linking / Mass: 133.103 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO4 Mass: 18.015 Da / Num. of mol.: 280 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 280 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL-5A / Wavelength: 1 Å
/ Beamline: BL-5A / Wavelength: 1 Å Processing
Processing