 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4z5d | ||||||
|---|---|---|---|---|---|---|---|
| Title | HipB-O4 21mer complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Transcription/DNA / HTH motif / Transcription repressor / HipA sequestering / Transcription-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtoxin-antitoxin complex / regulation of growth / DNA-binding transcription repressor activity / core promoter sequence-specific DNA binding / protein-DNA complex / sequence-specific DNA binding / transcription cis-regulatory region binding / negative regulation of DNA-templated transcription / DNA-templated transcription / regulation of DNA-templated transcription / protein homodimerization activity Similarity search - Function | ||||||
| Biological species |  synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.15 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.15 Å | ||||||
 Authors Authors | Min, J. / Brennan, R.G. / Schumacher, M.A. | ||||||
 Citation Citation | Journal: To be published Title: Molecular mechanism on hipBA gene regulation. Authors: Min, J. / Wang, A. / Brennan, R.G. / Schumacher, M.A. | ||||||
| History |
|
- Structure visualization
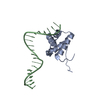
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4z5d.cif.gz | 72.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4z5d.ent.gz | 48.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4z5d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4z5d_validation.pdf.gz | 434.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4z5d_full_validation.pdf.gz | 435.1 KB | Display | |
| Data in XML | 4z5d_validation.xml.gz | 11.6 KB | Display | |
| Data in CIF | 4z5d_validation.cif.gz | 17.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/z5/4z5dftp://data.pdbj.org/pub/pdb/validation_reports/z5/4z5d | HTTPS FTP |
-Related structure data
| Related structure data |  4z58C 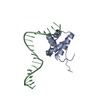 4z59C  4z5cC  4z5hC  4z52 4z56 4z57 4z5a 4z5b 4z5e 4z5f 4z5g 4z5i 4z5j 4z5k 4z5l 4z5m 4z5n 4z5o C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 8060.239 Da / Num. of mol.: 2 / Fragment: UNP residues 4-74 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: DNA chain | | Mass: 6438.172 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: DNA chain | | Mass: 6124.965 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 270 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 270 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.51 Å3/Da / Density % sol: 64.99 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 0.02M MgCl2, 0.05M sodium cacodylate trihydrate, pH 6.0, 5~30% 2-propanol, 0.001M Spermine |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E+ SUPERBRIGHT / Wavelength: 1.5418 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Jan 4, 2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Rigaku Varimax / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.15→50 Å / Num. obs: 22157 / % possible obs: 99.9 % / Redundancy: 7.1 % / Rmerge(I) obs: 0.048 / Χ2: 1.05 / Net I/av σ(I): 44.633 / Net I/σ(I): 15.8 / Num. measured all: 157548 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 4Z52 4z52 Resolution: 2.15→47.027 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.8872 / SU ML: 0.16 / Cross valid method: FREE R-VALUE / σ(F): 0.23 / Phase error: 18.09 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 73.57 Å2 / Biso mean: 26.0159 Å2 / Biso min: 10.32 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.15→47.027 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
|
