 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4xnu: X-ray structure of Drosophila dopamine transporter in complex wit... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xnu | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of Drosophila dopamine transporter in complex with nisoxetine | ||||||
 Components Components |
| ||||||
 Keywords Keywords | transport protein/inhibitor / integral membrane protein / all-alpha helical antidepressant complex / transport protein-inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / SLC-mediated transport of neurotransmitters / circadian sleep/wake cycle / cocaine binding / response to odorant / : / dopamine:sodium symporter activity / regulation of presynaptic cytosolic calcium ion concentration / : / sleep ...: / SLC-mediated transport of neurotransmitters / circadian sleep/wake cycle / cocaine binding / response to odorant / : / dopamine:sodium symporter activity / regulation of presynaptic cytosolic calcium ion concentration / : / sleep / neuronal cell body membrane / dopamine uptake involved in synaptic transmission / amino acid transport / sodium ion transmembrane transport / adult locomotory behavior / presynaptic membrane / axon / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |   | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å | ||||||
 Authors Authors | Aravind, P. / Wang, K. / Gouaux, E. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2015 Title: X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Authors: Penmatsa, A. / Wang, K.H. / Gouaux, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xnu.cif.gz | 204.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xnu.ent.gz | 156.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4xnu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xn/4xnuftp://data.pdbj.org/pub/pdb/validation_reports/xn/4xnu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4xnxC  4m48S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 59684.055 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  Homo sapiens (human) / References: UniProt: Q7K4Y6*PLUS Homo sapiens (human) / References: UniProt: Q7K4Y6*PLUS |
|---|
-Antibody , 2 types, 2 molecules LH
| #2: Antibody | Mass: 23306.586 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #3: Antibody | Mass: 25921.338 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
-Non-polymers , 5 types, 13 molecules 

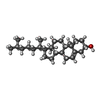


| #4: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Chemical | ChemComp-CL / |  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#6: Chemical | ChemComp-CLR / |  Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O#7: Chemical | ChemComp-41U / |  Mass: 271.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21NO2 Mass: 271.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21NO2#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.92 Å3/Da / Density % sol: 75.02 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / Details: PEG 350 MME 38%, Glycine 0.1M / PH range: 9 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 0.979 Å / Beamline: 24-ID-C / Wavelength: 0.979 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Mar 15, 2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.98→50 Å / Num. obs: 44010 / % possible obs: 99.4 % / Redundancy: 5.1 % / Biso Wilson estimate: 92.53 Å2 / Rmerge(I) obs: 0.133 / Rpim(I) all: 0.069 / Rrim(I) all: 0.137 / Χ2: 0.629 / Net I/av σ(I): 12.857 / Net I/σ(I): 3.9 / Num. measured all: 223836 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4M48 Resolution: 2.98→47.099 Å / FOM work R set: 0.7844 / SU ML: 0.4 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 27.21 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 177.48 Å2 / Biso mean: 85.68 Å2 / Biso min: 43.24 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.98→47.099 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
|
