 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4xjp: Crystal structure of 7,8-diaminopelargonic acid synthase (BioA) f... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xjp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of 7,8-diaminopelargonic acid synthase (BioA) from Mycobacterium tuberculosis, complexed with an inhibitor optimized from HTS lead | ||||||
 Components Components | Adenosylmethionine-8-amino-7-oxononanoate aminotransferase | ||||||
 Keywords Keywords | transferase/transferase inhibitor / Inhibitor Complex Transaminase PLP / transferase-transferase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationadenosylmethionine-8-amino-7-oxononanoate transaminase / S-adenosyl-L-methionine:8-amino-7-oxononanoate transaminase activity / biotin biosynthetic process / pyridoxal phosphate binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Finzel, B.C. / Dai, R. | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2017 Title: Structure-Based Optimization of Pyridoxal 5'-Phosphate-Dependent Transaminase Enzyme (BioA) Inhibitors that Target Biotin Biosynthesis in Mycobacterium tuberculosis. Authors: Liu, F. / Dawadi, S. / Maize, K.M. / Dai, R. / Park, S.W. / Schnappinger, D. / Finzel, B.C. / Aldrich, C.C. #1: Journal: THESIS, UNIVERSITY OF MINNESOTA / Year: 2015Title: FRAGMENT BASED INHIBITOR DESIGN OF MYCOBACTERIUM TUBERCULOSIS BIOA (HTTP://HDL.HANDLE.NET/11299/171084) Authors: Dai, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xjp.cif.gz | 201.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xjp.ent.gz | 156.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4xjp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xj/4xjpftp://data.pdbj.org/pub/pdb/validation_reports/xj/4xjp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4xjoC  5kgsC  5kgtC  3tftS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 48536.520 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (bacteria)Strain: ATCC 25618 / H37Rv / Gene: bioA, Rv1568, MTCY336.35c / Production host: References: UniProt: P9WQ81, adenosylmethionine-8-amino-7-oxononanoate transaminase |
|---|
-Non-polymers , 5 types, 890 molecules 
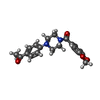



| #2: Chemical |  Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#3: Chemical |  Mass: 352.384 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H20N2O4 Mass: 352.384 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H20N2O4#4: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical |  Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 879 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.11 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 25 mM HEPES pH 7.5, 50 mM NaCl, 0.1 mM TCEP Reservoir:10% PEG 8000, 0.1M magnesium chloride, 0.1M HEPES pH 7.5 Cryo: 15% PEG 400 in reservoir solution |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å | |||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 21, 2014 | |||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||||||||
| Reflection | Resolution: 1.6→204.252 Å / Num. obs: 113574 / % possible obs: 99.7 % / Redundancy: 6.4 % / Rmerge(I) obs: 0.097 / Net I/σ(I): 13.3 / Num. measured all: 729227 | |||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3TFT Resolution: 1.6→32.539 Å / SU ML: 0.15 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 17.2 / Stereochemistry target values: ML
| ||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||
| Displacement parameters | Biso max: 72.23 Å2 / Biso mean: 17.7722 Å2 / Biso min: 5.14 Å2 | ||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.6→32.539 Å
|
