| Entry | Database: PDB / ID: 4r5r
|
|---|
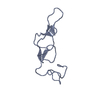
| Title | Crystal structure of Rhodostomin KKKRT mutant |
|---|
 Components Components | Disintegrin rhodostomin |
|---|
 Keywords Keywords | TOXIN / RGD motif / disintegrin / integrin / rhodostomin / linker region |
|---|
| Function / homology |  Function and homology information Function and homology information
Hydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases / metalloendopeptidase activity / toxin activity / proteolysis / extracellular region / metal ion binding / plasma membraneSimilarity search - Function Disintegrin domain / Echistatin / Disintegrin, conserved site / Disintegrins signature. / Peptidase M12B, propeptide / Reprolysin family propeptide / Reprolysin domain, adamalysin-type / Reprolysin (M12B) family zinc metalloprotease / Disintegrin / Disintegrin domain profile. ...Disintegrin domain / Echistatin / Disintegrin, conserved site / Disintegrins signature. / Peptidase M12B, propeptide / Reprolysin family propeptide / Reprolysin domain, adamalysin-type / Reprolysin (M12B) family zinc metalloprotease / Disintegrin / Disintegrin domain profile. / Homologues of snake disintegrins / Disintegrin domain / Disintegrin domain superfamily / Peptidase M12B, ADAM/reprolysin / ADAM type metalloprotease domain profile. / Metallopeptidase, catalytic domain superfamily / Few Secondary Structures / Irregular / Neutral zinc metallopeptidases, zinc-binding region signature.Similarity search - Domain/homology |
|---|
| Biological species |  Calloselasma rhodostoma (Malayan pit viper) Calloselasma rhodostoma (Malayan pit viper) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.96 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.96 Å |
|---|
 Authors Authors | Huang, C.H. / Shiu, J.H. / Chang, Y.T. / Jeng, W.Y. / Chuang, W.J. |
|---|
 Citation Citation | Journal: To be Published
Title: Effects of the regions adjacent to the RGD motif in disintegrins on their inhibitory activities and structures
Authors: Huang, C.H. / Shiu, J.H. / Chang, Y.T. / Jeng, W.Y. / Chuang, W.J. |
|---|
| History | | Deposition | Aug 21, 2014 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Aug 26, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 22, 2017 | Group: Refinement description / Category: software |
|---|
| Revision 1.2 | Oct 30, 2024 | Group: Data collection / Database references / Structure summary
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature / struct_ref_seq_dif
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj



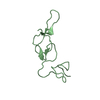
 Assembly
Assembly

 Pichia pastoris (fungus) / References: UniProt: P30403
Pichia pastoris (fungus) / References: UniProt: P30403 Mass: 18.015 Da / Num. of mol.: 264 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 264 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL13B1 / Wavelength: 0.97622 Å
/ Beamline: BL13B1 / Wavelength: 0.97622 Å Processing
Processing