 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
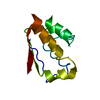
Yorodumi- PDB-4q6h: CFTR Associated Ligand (CAL) bound to last 6 residues of CFTR (de... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4q6h | ||||||
|---|---|---|---|---|---|---|---|
| Title | CFTR Associated Ligand (CAL) bound to last 6 residues of CFTR (decameric peptide: iCAL36VQDTRL) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSPORT PROTEIN / PDZ-peptide complex / cystic fibrosis transmembrane conductance regulator | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of anion channel activity / RHO GTPases regulate CFTR trafficking / negative regulation of protein localization to cell surface / Golgi to plasma membrane transport / trans-Golgi network transport vesicle / Golgi-associated vesicle membrane / apical protein localization / RHOQ GTPase cycle / molecular sequestering activity / endoplasmic reticulum to Golgi vesicle-mediated transport ...negative regulation of anion channel activity / RHO GTPases regulate CFTR trafficking / negative regulation of protein localization to cell surface / Golgi to plasma membrane transport / trans-Golgi network transport vesicle / Golgi-associated vesicle membrane / apical protein localization / RHOQ GTPase cycle / molecular sequestering activity / endoplasmic reticulum to Golgi vesicle-mediated transport / protein transport / transmembrane transporter binding / postsynaptic density / Golgi membrane / lysosomal membrane / dendrite / Golgi apparatus / protein-containing complex / membrane / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.903 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.903 Å | ||||||
 Authors Authors | Amacher, J.F. / Madden, D.R. | ||||||
 Citation Citation | Journal: TO BE PUBLISHED Title: Understanding PDZ Affinity and Selectivity: All Residues Considered Authors: Amacher, J.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4q6h.cif.gz | 48.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4q6h.ent.gz | 34.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4q6h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q6/4q6hftp://data.pdbj.org/pub/pdb/validation_reports/q6/4q6h | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4e34S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 9353.722 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GOPC, CAL, FIG / Production host:  |
|---|---|
| #2: Protein/peptide | Mass: 731.818 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: peptide synthesis |
| #3: Chemical | ChemComp-SO3 / 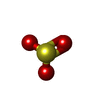  Mass: 80.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO3 Mass: 80.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO3 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 51 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 51 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.59 Å3/Da / Density % sol: 52.52 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 37% (w/v) polyethylene glycol (PEG) 1000, 0.07 M sodium thiosulfate pentahydrate, 0.1 M Tris pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X6A / Wavelength: 1.0781 Å / Beamline: X6A / Wavelength: 1.0781 Å | |||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Nov 8, 2013 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: S1 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.0781 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.9→19.123 Å / Num. all: 9142 / Num. obs: 9041 / % possible obs: 98.9 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 9.08 / Rsym value: 0.085 / Net I/σ(I): 48.39 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ID 4E34 (CAL PDZ, no peptide included in search model) Resolution: 1.903→19.123 Å / SU ML: 0.12 / σ(F): 1.36 / σ(I): 9.08 / Phase error: 20.18 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 48.784 Å2 / ksol: 0.431 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.903→19.123 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 10.4388 Å / Origin y: 22.9653 Å / Origin z: 36.4886 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: chain A and resid 284:370 |
