 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4lz3: F95H Epi-isozizaene synthase: Complex with Mg, inorganic pyrophos... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4lz3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | F95H Epi-isozizaene synthase: Complex with Mg, inorganic pyrophosphate and benzyl triethyl ammonium cation | ||||||
 Components Components | Epi-isozizaene synthase | ||||||
 Keywords Keywords | LYASE / Class I terpene cyclase | ||||||
| Function / homology |  Function and homology information Function and homology informationepi-isozizaene synthase / epi-isozizaene synthase activity / terpene synthase activity / metal ion binding Similarity search - Function | ||||||
| Biological species |  Streptomyces coelicolor (bacteria) Streptomyces coelicolor (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.095 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.095 Å | ||||||
 Authors Authors | Li, R. / Chou, W. / Himmelberger, J.A. / Litwin, K. / Harris, G. / Cane, D.E. / Christianson, D.W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2014 Title: Reprogramming the Chemodiversity of Terpenoid Cyclization by Remolding the Active Site Contour of epi-Isozizaene Synthase. Authors: Li, R. / Chou, W.K. / Himmelberger, J.A. / Litwin, K.M. / Harris, G.G. / Cane, D.E. / Christianson, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4lz3.cif.gz | 94.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4lz3.ent.gz | 68.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4lz3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lz/4lz3ftp://data.pdbj.org/pub/pdb/validation_reports/lz/4lz3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ltvC  4ltzC  4luuC  4lxwC  4lz0C  4lzcC  3kb9S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 43706.984 Da / Num. of mol.: 1 / Mutation: F95H Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces coelicolor (bacteria) / Strain: A3(2) / Gene: cyc1, SCO5222, SC7E4.19 / Plasmid: pET28a(+) / Production host: |
|---|
-Non-polymers , 6 types, 278 molecules 

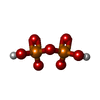


| #2: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical |  Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-POP / |  Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2#5: Chemical | ChemComp-BTM / |  Mass: 192.320 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H22N Mass: 192.320 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H22N#6: Chemical | ChemComp-PI / |  Mass: 95.979 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO4P Mass: 95.979 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO4P#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Nonpolymer details | At pH 7.5, theoretically, both dihydrogen phosphate and hydrogen phosphate ions are present with a ...At pH 7.5, theoretically, both dihydrogen phosphate and hydrogen phosphate ions are present with a ratio around 1:2. In the crystal structure of F95H EIZS, the dihydrogen phosphate /hydrogen phosphate ion is surrounded and stabilized by three Arginine side chains, one Histidine side chain, one glycerol and two water molecules. The contacts involving hydrogen bonding interaction and salt bridge interaction between each oxygen of dihydrogen phosphate /hydrogen phosphate ion and surrounding molecules indicates that hydrogen phosphate ion would be a reasonable fit in such electropositive environment. |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.41 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: A 0.6 uL drop of protein solution [10 mg/mL F95H EIZS, 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM TCEP, 2 mM sodium pyrophosphate, 2 mM BTAC] was added to 0.6 uL ...Details: A 0.6 uL drop of protein solution [10 mg/mL F95H EIZS, 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM TCEP, 2 mM sodium pyrophosphate, 2 mM BTAC] was added to 0.6 uL of precipitant solution [40 mM KH2PO4, 16% polyethylene glycol 8000, and 20% glycerol] and equilibrated against a 110 uL well reservoir of precipitant solution., VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X29A / Wavelength: 1.075 Å / Beamline: X29A / Wavelength: 1.075 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Feb 2, 2013 Details: Monochromator: Double silicon (111) crystal monochromator with cryogenically-cooled first crystal and sagittally-bent second crystal horizontally-focusing at 3.3:1 demagnification. Mirror: ...Details: Monochromator: Double silicon (111) crystal monochromator with cryogenically-cooled first crystal and sagittally-bent second crystal horizontally-focusing at 3.3:1 demagnification. Mirror: Meridionally-bent fused silica mirror with palladium and uncoated stripes vertically focusing at 6.6:1 demagnification. |
| Radiation | Monochromator: Double silicon (111) crystal monochromator with cryogenically-cooled first crystal and sagittally-bent second crystal horizontally-focusing at 3.3:1 demagnification. Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.075 Å / Relative weight: 1 |
| Reflection | Resolution: 2.095→50 Å / Num. obs: 23067 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 5.4 % / Rmerge(I) obs: 0.136 / Rsym value: 0.136 / Net I/σ(I): 11.859 |
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.323 / Mean I/σ(I) obs: 4.917 / Rsym value: 0.323 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDBID: 3KB9 Resolution: 2.095→43.829 Å / SU ML: 0.17 / σ(F): 1.35 / Phase error: 18.94 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.095→43.829 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
