- PDB-4kax: Crystal structure of the Grp1 PH domain in complex with Arf6-GTP -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4kax
Title
Crystal structure of the Grp1 PH domain in complex with Arf6-GTP
Components
ADP-ribosylation factor 6
Cytohesin-3
Keywords
PROTEIN BINDING/SIGNALING PROTEIN / PH domain / Phosphoinositides / PROTEIN BINDING-SIGNALING PROTEIN complex
Function / homology
Function and homology information
maintenance of postsynaptic density structure / protein localization to cleavage furrow / establishment of epithelial cell polarity / positive regulation of mitotic cytokinetic process / Golgi vesicle transport / regulation of dendritic spine development / Intra-Golgi traffic / negative regulation of protein localization to cell surface / protein localization to endosome / regulation of Rac protein signal transduction ...maintenance of postsynaptic density structure / protein localization to cleavage furrow / establishment of epithelial cell polarity / positive regulation of mitotic cytokinetic process / Golgi vesicle transport / regulation of dendritic spine development / Intra-Golgi traffic / negative regulation of protein localization to cell surface / protein localization to endosome / regulation of Rac protein signal transduction / negative regulation of dendrite development / ruffle assembly / regulation of filopodium assembly / positive regulation of keratinocyte migration / positive regulation of focal adhesion disassembly / MET receptor recycling / endocytic recycling / thioesterase binding / regulation of ARF protein signal transduction / Flemming body / filopodium membrane / TBC/RABGAPs / cortical actin cytoskeleton organization / protein localization to cell surface / positive regulation of actin filament polymerization / cleavage furrow / phosphatidylinositol-3,4,5-trisphosphate binding / bicellular tight junction / endocytic vesicle / regulation of presynapse assembly / synaptic vesicle endocytosis / ruffle / vesicle-mediated transport / signaling adaptor activity / positive regulation of cell adhesion / guanyl-nucleotide exchange factor activity / protein localization to plasma membrane / positive regulation of protein secretion / small monomeric GTPase / positive regulation of protein localization to plasma membrane / adherens junction / positive regulation of neuron projection development / negative regulation of receptor-mediated endocytosis / cellular response to nerve growth factor stimulus / intracellular protein transport / recycling endosome membrane / GDP binding / nervous system development / Clathrin-mediated endocytosis / presynapse / G protein activity / early endosome membrane / midbody / cell cortex / cell differentiation / cell adhesion / postsynapse / endosome / Golgi membrane / cell division / focal adhesion / GTPase activity / GTP binding / glutamatergic synapse / Golgi apparatus / extracellular exosome / nucleoplasm / membrane / plasma membrane / cytosol / cytoplasm Similarity search - Function
ADP-ribosylation factor 6 / Sec7 domain / Sec7, C-terminal domain superfamily / Sec7 domain superfamily / Sec7 domain / SEC7 domain profile. / Sec7 domain / Small GTPase superfamily, ARF type / Small GTPase Arf domain profile. / Sar1p-like members of the Ras-family of small GTPases ...ADP-ribosylation factor 6 / Sec7 domain / Sec7, C-terminal domain superfamily / Sec7 domain superfamily / Sec7 domain / SEC7 domain profile. / Sec7 domain / Small GTPase superfamily, ARF type / Small GTPase Arf domain profile. / Sar1p-like members of the Ras-family of small GTPases / Small GTPase superfamily, ARF/SAR type / ADP-ribosylation factor family / ARF-like small GTPases; ARF, ADP-ribosylation factor / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / PH domain / PH domain profile. / Pleckstrin homology domain. / Pleckstrin homology domain / Rab subfamily of small GTPases / Small GTP-binding protein domain / PH-like domain superfamily / Roll / P-loop containing nucleotide triphosphate hydrolases / Rossmann fold / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Mainly Beta / Alpha Beta Similarity search - Domain/homology
Mass: 19530.291 Da / Num. of mol.: 1 / Fragment: Arf6 (residues 14-181) / Mutation: Q67L Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ARF6 / Production host: Escherichia coli (E. coli) / References: UniProt: P62330
#2: Protein
Cytohesin-3 / ARF nucleotide-binding site opener 3 / Protein ARNO3 / General receptor of phosphoinositides 1 / ...ARF nucleotide-binding site opener 3 / Protein ARNO3 / General receptor of phosphoinositides 1 / Grp1 / PH / SEC7 and coiled-coil domain-containing protein 3
Mass: 19038.633 Da / Num. of mol.: 1 / Fragment: Grp1 PH domain (residues 247-399) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CYTH3, ARNO3, GRP1, PSCD3 / Production host: Escherichia coli (E. coli) / References: UniProt: O43739
Mass: 18.015 Da / Num. of mol.: 426 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.85 Å3/Da / Density % sol: 56.81 %
Crystal grow
Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 8.8 Details: 50 mM Tris, 20% PEG 4000, 0.2 M sodium citrate. Microseeding and TCEP were needed for large crystal growth, pH 8.8, VAPOR DIFFUSION, HANGING DROP, temperature 291K
Monochromator: Double silicon(111) crystal monochromator with cryogenically-cooled first crystal and sagittally-bent second crystal horizontally-focusing at 3.3:1 demagnification Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj




 Assembly
Assembly






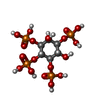

 Mass: 523.180 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM
Mass: 523.180 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7
Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 500.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H16O18P4
Mass: 500.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H16O18P4 Sample preparation
Sample preparation / Beamline: X25 / Wavelength: 1.1
/ Beamline: X25 / Wavelength: 1.1  Processing
Processing