 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4jx7: Crystal structure of Pim1 kinase in complex with inhibitor 2-[(tr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4jx7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Pim1 kinase in complex with inhibitor 2-[(trans-4-aminocyclohexyl)amino]-4-{[3-(trifluoromethyl)phenyl]amino}pyrido[4,3-d]pyrimidin-5(6H)-one | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / Serine/threonine-protein kinases / ATP binding / Phosphorylation / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / : / positive regulation of protein serine/threonine kinase activity ...regulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / : / positive regulation of protein serine/threonine kinase activity / ribosomal small subunit binding / positive regulation of cardiac muscle cell proliferation / positive regulation of brown fat cell differentiation / positive regulation of TORC1 signaling / Signaling by FLT3 fusion proteins / negative regulation of innate immune response / regulation of mitotic cell cycle / protein serine/threonine kinase activator activity / cellular response to type II interferon / manganese ion binding / protein autophosphorylation / Interleukin-4 and Interleukin-13 signaling / protein phosphorylation / non-specific serine/threonine protein kinase / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / apoptotic process / nucleolus / negative regulation of apoptotic process / positive regulation of DNA-templated transcription / nucleoplasm / ATP binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Lee, S.J. / Han, B.G. / Cho, J.W. / Choi, J.S. / Lee, J.K. / Song, H.J. / Koh, J.S. / Lee, B.I. | ||||||
 Citation Citation | Journal: Plos One / Year: 2013 Title: Crystal structure of pim1 kinase in complex with a pyrido[4,3-d]pyrimidine derivative suggests a unique binding mode. Authors: Lee, S.J. / Han, B.G. / Cho, J.W. / Choi, J.S. / Lee, J. / Song, H.J. / Koh, J.S. / Lee, B.I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4jx7.cif.gz | 74.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4jx7.ent.gz | 54.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4jx7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jx/4jx7ftp://data.pdbj.org/pub/pdb/validation_reports/jx/4jx7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4jx3C  1xqzS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37293.410 Da / Num. of mol.: 1 / Fragment: SEE REMARK 999 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PIM1 / Plasmid: pET32 / Production host: References: UniProt: P11309, non-specific serine/threonine protein kinase |
|---|---|
| #2: Protein/peptide | Mass: 1592.850 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #3: Chemical | ChemComp-1N6 / 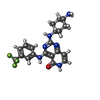  Mass: 418.416 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H21F3N6O Mass: 418.416 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H21F3N6O |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 104 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 104 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | PIM-1 IS ISOFORM 2 (UNP P11309-2, RESIDUES 92-404). |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.87 Å3/Da / Density % sol: 57.16 % |
|---|---|
| Crystal grow | Temperature: 297 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 0.7 M sodium potassium tartrate, 0.1 M MES, pH 6.5, VAPOR DIFFUSION, SITTING DROP, temperature 297K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS  / Beamline: 6C1 / Wavelength: 1.2399 Å / Beamline: 6C1 / Wavelength: 1.2399 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jan 13, 2010 |
| Radiation | Monochromator: double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.2399 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. obs: 16996 / % possible obs: 98.3 % |
| Reflection shell | Resolution: 2.4→2.49 Å / % possible all: 99.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1XQZ Resolution: 2.4→50 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.916 / SU B: 6.118 / SU ML: 0.144 / Cross valid method: THROUGHOUT / ESU R: 0.286 / ESU R Free: 0.217 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.229 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.463 Å / Total num. of bins used: 20
|
