 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4jd6: Crystal structure of Mycobacterium tuberculosis Eis in complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4jd6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Mycobacterium tuberculosis Eis in complex with coenzyme A and tobramycin | ||||||
 Components Components | Enhanced intracellular survival protein | ||||||
 Keywords Keywords | TRANSFERASE / GNAT / aminoglycoside acetyltransferase | ||||||
| Function / homology |  Function and homology information Function and homology informationeffector-mediated defense to host-produced reactive oxygen species / symbiont-mediated perturbation of host inflammatory response / symbiont-mediated perturbation of host innate immune response / symbiont-mediated suppression of host programmed cell death / Suppression of autophagy / aminoglycoside antibiotic catabolic process / aminoglycoside N-acetyltransferase activity / bacterial extracellular vesicle / symbiont-mediated perturbation of host programmed cell death / biological process involved in interaction with host ...effector-mediated defense to host-produced reactive oxygen species / symbiont-mediated perturbation of host inflammatory response / symbiont-mediated perturbation of host innate immune response / symbiont-mediated suppression of host programmed cell death / Suppression of autophagy / aminoglycoside antibiotic catabolic process / aminoglycoside N-acetyltransferase activity / bacterial extracellular vesicle / symbiont-mediated perturbation of host programmed cell death / biological process involved in interaction with host / N-acetyltransferase activity / host cell cytoplasmic vesicle / protein-lysine-acetyltransferase activity / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / host extracellular region / response to antibiotic / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å | ||||||
 Authors Authors | Biswas, T. / Chen, W. / Garneau-Tsodikova, S. / Tsodikov, O.V. | ||||||
 Citation Citation | Journal: Chembiochem / Year: 2013 Title: Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase eis. Authors: Houghton, J.L. / Biswas, T. / Chen, W. / Tsodikov, O.V. / Garneau-Tsodikova, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4jd6.cif.gz | 458.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4jd6.ent.gz | 379.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4jd6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jd/4jd6ftp://data.pdbj.org/pub/pdb/validation_reports/jd/4jd6 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 46660.840 Da / Num. of mol.: 6 / Mutation: C204A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Strain: H37Rv / Gene: RVBD_2416c / Production host: #2: Chemical | ChemComp-COA / 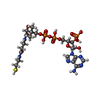  Mass: 767.534 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C21H36N7O16P3S Mass: 767.534 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C21H36N7O16P3S#3: Chemical |   Mass: 467.514 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H37N5O9 / Comment: antibiotic*YM Mass: 467.514 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H37N5O9 / Comment: antibiotic*YMNonpolymer details | The phosphopantetheinyl arms of CoA molecules were poorly resolved in chains A, D, E, F and were ...The phosphopantetheinyl arms of CoA molecules were poorly resolved in chains A, D, E, F and were placed in the partial electron density based on their conformation in the other monomers and consistent with the previously observed position of the phosphopantetheinyl arm of CoA molecules mound to Eis in entry 3R1K. Similarly, the tobramycin molecule bound to monomer B was placed and refined in partial, but strong, omit density based on the position of the tobramycin molecule in the binding site of monomer A. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.51 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: TOB (1 mM) and CoA (1 mM)); reservoir solution (Tris-HCl (100 mM, pH 8.5) and PEG 8,000 (13% w/v)), VAPOR DIFFUSION, HANGING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-D / Wavelength: 1 Å / Beamline: 21-ID-D / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Nov 11, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→50 Å / Num. obs: 33556 / % possible obs: 100 % / Observed criterion σ(I): 2.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 3.5→40 Å / Cor.coef. Fo:Fc: 0.927 / Cor.coef. Fo:Fc free: 0.901 / SU B: 42.469 / SU ML: 0.626 / Cross valid method: THROUGHOUT / ESU R Free: 0.733 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 126.345 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.5→3.59 Å / Total num. of bins used: 20
|
