- PDB-4j9u: Crystal Structure of the TrkH/TrkA potassium transport complex -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4j9u
Title
Crystal Structure of the TrkH/TrkA potassium transport complex
Components
Potassium uptake protein TrkA
Trk system potassium uptake protein TrkH
Keywords
TRANSPORT PROTEIN / RCK domain / potassium transport / membrane protein / Structural Genomics / PSI-Biology / New York Consortium on Membrane Protein Structure / NYCOMPS
Function / homology
Function and homology information
potassium:chloride symporter activity / potassium ion transmembrane transporter activity / potassium ion binding / potassium channel activity / potassium ion transmembrane transport / nucleotide binding / protein homodimerization activity / metal ion binding / identical protein binding / plasma membrane Similarity search - Function
Potassium uptake protein TrkA / TrkH potassium transport family / Cation transporter / Cation transport protein / Regulator of K+ conductance, C-terminal domain / : / TrkA-N domain / Regulator of K+ conductance, C-terminal / Regulator of K+ conductance, C-terminal domain superfamily / TrkA-C domain ...Potassium uptake protein TrkA / TrkH potassium transport family / Cation transporter / Cation transport protein / Regulator of K+ conductance, C-terminal domain / : / TrkA-N domain / Regulator of K+ conductance, C-terminal / Regulator of K+ conductance, C-terminal domain superfamily / TrkA-C domain / RCK C-terminal domain profile. / Regulator of K+ conductance, N-terminal / RCK N-terminal domain profile. / NAD(P)-binding Rossmann-like Domain / Alpha-Beta Plaits / NAD(P)-binding domain superfamily / Rossmann fold / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
: / NICOTINAMIDE-ADENINE-DINUCLEOTIDE / HEXATANTALUM DODECABROMIDE / Trk system potassium uptake protein TrkA / Trk system potassium uptake protein TrkH Similarity search - Component
Biological species
Vibrio parahaemolyticus (bacteria)
Method
X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 3.8 Å
A: Trk system potassium uptake protein TrkH B: Trk system potassium uptake protein TrkH C: Trk system potassium uptake protein TrkH D: Trk system potassium uptake protein TrkH E: Potassium uptake protein TrkA F: Potassium uptake protein TrkA G: Potassium uptake protein TrkA H: Potassium uptake protein TrkA hetero molecules
A: Trk system potassium uptake protein TrkH B: Trk system potassium uptake protein TrkH E: Potassium uptake protein TrkA F: Potassium uptake protein TrkA G: Potassium uptake protein TrkA H: Potassium uptake protein TrkA hetero molecules
C: Trk system potassium uptake protein TrkH D: Trk system potassium uptake protein TrkH E: Potassium uptake protein TrkA F: Potassium uptake protein TrkA G: Potassium uptake protein TrkA H: Potassium uptake protein TrkA hetero molecules
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Vibrio parahaemolyticus (bacteria)
Vibrio parahaemolyticus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj

 Assembly
Assembly


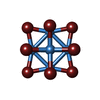
 Mass: 2044.535 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: Br12Ta6
Mass: 2044.535 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: Br12Ta6
 Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K
 Mass: 663.425 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM
Mass: 663.425 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Sample preparation
Sample preparation / Beamline: X25 / Wavelength: 1.2515 Å
/ Beamline: X25 / Wavelength: 1.2515 Å Processing
Processing