 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4i0h: SPR and structural analysis yield insight towards mechanism of in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4i0h | ||||||
|---|---|---|---|---|---|---|---|
| Title | SPR and structural analysis yield insight towards mechanism of inhibition of BACE inhibitors. | ||||||
 Components Components | Beta-secretase 1 | ||||||
 Keywords Keywords | hydrolase/hydrolase inhibitor / BACE / Asp protease / hydrolysis / hydrolase-hydrolase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationmemapsin 2 / Golgi-associated vesicle lumen / beta-aspartyl-peptidase activity / signaling receptor ligand precursor processing / amyloid-beta formation / amyloid precursor protein catabolic process / membrane protein ectodomain proteolysis / amyloid-beta metabolic process / detection of mechanical stimulus involved in sensory perception of pain / response to insulin-like growth factor stimulus ...memapsin 2 / Golgi-associated vesicle lumen / beta-aspartyl-peptidase activity / signaling receptor ligand precursor processing / amyloid-beta formation / amyloid precursor protein catabolic process / membrane protein ectodomain proteolysis / amyloid-beta metabolic process / detection of mechanical stimulus involved in sensory perception of pain / response to insulin-like growth factor stimulus / prepulse inhibition / cellular response to manganese ion / multivesicular body / swimming behavior / cellular response to copper ion / presynaptic modulation of chemical synaptic transmission / hippocampal mossy fiber to CA3 synapse / protein serine/threonine kinase binding / trans-Golgi network / protein processing / recycling endosome / response to lead ion / cellular response to amyloid-beta / synaptic vesicle / late endosome / peptidase activity / positive regulation of neuron apoptotic process / amyloid-beta binding / endopeptidase activity / amyloid fibril formation / aspartic-type endopeptidase activity / early endosome / lysosome / endosome / endosome membrane / membrane raft / endoplasmic reticulum lumen / Amyloid fiber formation / axon / neuronal cell body / dendrite / enzyme binding / cell surface / Golgi apparatus / proteolysis / membrane / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Yao, N. / Brecht, E. | ||||||
 Citation Citation | Journal: To be Published Title: SPR and structural analysis yield insight towards mechanism of inhibition of BACE inhibitors Authors: Mondal, K. / Regnstrom, K. / Morishige, W. / Barbour, R. / Beroza, P. / Yao, N. / Bova, M.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4i0h.cif.gz | 238.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4i0h.ent.gz | 192.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4i0h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i0/4i0hftp://data.pdbj.org/pub/pdb/validation_reports/i0/4i0h | HTTPS FTP |
|---|
-Related structure data


















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
| #1: Protein | Mass: 45445.121 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BACE1, BACE, KIAA1149 / Production host:  #2: Chemical | 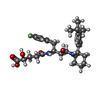  Mass: 560.672 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C31H42F2N2O5 Mass: 560.672 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C31H42F2N2O5#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.12 Å3/Da / Density % sol: 60.51 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 5.3 Details: Human BACE protein containing residues 57-453 and a C-terminal 6His-tag was concentrated to 6mg/ml in buffer 0.1M borate pH 8.5. Compound was added to give a final molar access of compound: ...Details: Human BACE protein containing residues 57-453 and a C-terminal 6His-tag was concentrated to 6mg/ml in buffer 0.1M borate pH 8.5. Compound was added to give a final molar access of compound:protein of 8:1. Protein and compound were then incubated on ice for 1 hour. The drops were set up with a 1:1(v/v) ratio of protein to mother liquor in a total volume of 2 ul. Diffraction quality crystals of BACE in complex with compound was obtained by sitting-drop vapor diffusion method at 291K against a reservoir containing 0.1 M sodium acetate, 2% PEG8000, VAPOR DIFFUSION, SITTING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS HTC / Detector: IMAGE PLATE / Date: Jan 1, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→60 Å / Num. all: 84800 / Num. obs: 87435 / % possible obs: 97 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.2→60 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.933 / SU B: 7.136 / SU ML: 0.175 / Cross valid method: THROUGHOUT / ESU R: 0.226 / ESU R Free: 0.204 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 67.888 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→60 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 2870 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.257 Å / Total num. of bins used: 20
|
