 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hyt: Na,K-ATPase in the E2P state with bound ouabain and Mg2+ in the c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hyt | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Na,K-ATPase in the E2P state with bound ouabain and Mg2+ in the cation-binding site | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE/MEMBRANE PROTEIN / membrane transporter / HYDROLASE-MEMBRANE PROTEIN complex | |||||||||
| Function / homology |  Function and homology information Function and homology information: / Ion homeostasis / Ion transport by P-type ATPases / Na+/K+-exchanging ATPase / regulation of monoatomic ion transport / positive regulation of sodium ion export across plasma membrane / positive regulation of potassium ion import across plasma membrane / P-type sodium:potassium-exchanging transporter activity / sodium ion binding / sodium:potassium-exchanging ATPase complex ...: / Ion homeostasis / Ion transport by P-type ATPases / Na+/K+-exchanging ATPase / regulation of monoatomic ion transport / positive regulation of sodium ion export across plasma membrane / positive regulation of potassium ion import across plasma membrane / P-type sodium:potassium-exchanging transporter activity / sodium ion binding / sodium:potassium-exchanging ATPase complex / membrane repolarization / establishment or maintenance of transmembrane electrochemical gradient / sodium ion export across plasma membrane / regulation of calcium ion transmembrane transport / positive regulation of potassium ion transmembrane transport / ion channel regulator activity / intracellular sodium ion homeostasis / relaxation of cardiac muscle / regulation of cardiac muscle contraction by calcium ion signaling / positive regulation of sodium ion transmembrane transport / organelle membrane / potassium ion binding / potassium ion import across plasma membrane / ATPase activator activity / intracellular potassium ion homeostasis / intercalated disc / lateral plasma membrane / transporter activator activity / sperm flagellum / ATP metabolic process / regulation of sodium ion transport / cardiac muscle contraction / T-tubule / proton transmembrane transport / protein localization to plasma membrane / sarcolemma / transmembrane transport / intracellular calcium ion homeostasis / melanosome / ATPase binding / regulation of gene expression / basolateral plasma membrane / protein-macromolecule adaptor activity / cell adhesion / apical plasma membrane / protein stabilization / innate immune response / axon / protein kinase binding / ATP hydrolysis activity / ATP binding / membrane / plasma membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.404 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.404 Å | |||||||||
 Authors Authors | Laursen, M. / Yatime, L. / Nissen, P. / Fedosova, N.U. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2013 Title: Crystal structure of the high-affinity Na+,K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Authors: Laursen, M. / Yatime, L. / Nissen, P. / Fedosova, N.U. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hyt.cif.gz | 1.1 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hyt.ent.gz | 917.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4hyt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hy/4hytftp://data.pdbj.org/pub/pdb/validation_reports/hy/4hyt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3n23S S: Starting model for refinement |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
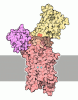





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Sodium/potassium-transporting ATPase subunit ... , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 112877.562 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) #2: Protein | Mass: 35204.402 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) |
|---|
-Protein , 1 types, 2 molecules GE
| #3: Protein | Mass: 7094.115 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) |
|---|
-Sugars , 2 types, 6 molecules 
| #4: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source |
|---|---|
| #12: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 5 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 5Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
-Non-polymers , 8 types, 27 molecules 

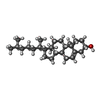





| #5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 584.652 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C29H44O12 Mass: 584.652 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C29H44O12#7: Chemical | ChemComp-CLR /  Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O#8: Chemical | ChemComp-1AT / |  Mass: 496.546 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H40O12 Mass: 496.546 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H40O12#9: Chemical | ChemComp-1DS / |  Mass: 496.546 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H40O12 Mass: 496.546 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H40O12#10: Chemical | ChemComp-17F /  Mass: 788.043 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C42H78NO10P / Comment: phospholipid*YM Mass: 788.043 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C42H78NO10P / Comment: phospholipid*YM#11: Chemical |  Mass: 538.755 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H58O9 Mass: 538.755 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H58O9#13: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.53 Å3/Da / Density % sol: 77.75 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: hanging drop / pH: 6.2 Details: 16-17% PEG 2000 mme, 10% Glycerol, 200 mM MgCl2, 100 mM MES-NMDG pH 6.2, 100 mM Urea, 5% tert-Butanol, 5mM DTT, 1.6 mM Sucrose monodecanoate, hanging drop, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Aug 14, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.4→54 Å / Num. all: 95443 / Num. obs: 72261 / % possible obs: 75.7 % / Observed criterion σ(F): 1.4 / Observed criterion σ(I): 1.4 / Redundancy: 6.7 % / Biso Wilson estimate: 111.5 Å2 / Rsym value: 0.072 / Net I/σ(I): 15.96 |
| Reflection shell | Resolution: 3.4→3.49 Å / Redundancy: 6.7 % / Mean I/σ(I) obs: 1.41 / Rsym value: 1.512 / % possible all: 4.8 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb ID 3N23 Resolution: 3.404→49.829 Å / Occupancy max: 1 / Occupancy min: 1 / SU ML: 0.38 / σ(F): 1.99 / Phase error: 26.15 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 128.844 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.404→49.829 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
