 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hld: Sulfonylpiperidines as Novel, Antibacterial Inhibitors of Gram-Po... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hld | ||||||
|---|---|---|---|---|---|---|---|
| Title | Sulfonylpiperidines as Novel, Antibacterial Inhibitors of Gram-Positive Thymidylate Kinase (TMK): Compound 11 | ||||||
 Components Components | Thymidylate kinase | ||||||
 Keywords Keywords | transferase/transferase inhibitor / TMK / kinase / thymidylate kinase / MRSA / pipiridine / transferase-transferase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationdTMP kinase / dTMP kinase activity / dUDP biosynthetic process / dTDP biosynthetic process / dTTP biosynthetic process / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus subsp. aureus (bacteria) Staphylococcus aureus subsp. aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Olivier, N. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2013 Title: Sulfonylpiperidines as novel, antibacterial inhibitors of Gram-positive thymidylate kinase (TMK). Authors: Martinez-Botella, G. / Loch, J.T. / Green, O.M. / Kawatkar, S.P. / Olivier, N.B. / Boriack-Sjodin, P.A. / Keating, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hld.cif.gz | 94.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hld.ent.gz | 72.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4hld.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4hld_validation.pdf.gz | 1.2 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4hld_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | 4hld_validation.xml.gz | 18.3 KB | Display | |
| Data in CIF | 4hld_validation.cif.gz | 25.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hl/4hldftp://data.pdbj.org/pub/pdb/validation_reports/hl/4hld | HTTPS FTP |
-Related structure data















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23454.586 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus subsp. aureus (bacteria)Strain: MRSA252 / Gene: SAR0483, tmk / Plasmid: pET30 / Production host: #2: Chemical | 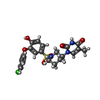  Mass: 491.945 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H22ClN3O6S Mass: 491.945 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H22ClN3O6S#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.55 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.8 Details: To obtain the inhibitor bound crystal form of TMK-S.aureus crystals were initially grown in the absence of compound using the sitting drop method at 293 K with a reservoir solution of 100 mM ...Details: To obtain the inhibitor bound crystal form of TMK-S.aureus crystals were initially grown in the absence of compound using the sitting drop method at 293 K with a reservoir solution of 100 mM PCPT (propionate-cacodylate-bistris propane buffer) pH 7-8, 21-24% PEG 3350, 200 mM Mg2Cl using 1:1 protein:reservoir solution with the protein solution at 13 mg/mL. Crystals were harvested and soaked overnight in a solution containing 100 mM PCPT, 35% PEG 3350, 200 mM Mg2Cl and 1-2 mM TK-666 from a 100 mM DMSO stock. After soaking the crystals were cryoprotected by soaking for 15 minutes in compound-soak solution supplemented with 20% ethylene glycol. Cryoprotected crystals were mounted on nylon loops and flash-cooled in liquid nitrogen., VAPOR DIFFUSION, SITTING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E+ SUPERBRIGHT / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN 944+ / Detector: CCD / Date: Nov 23, 2009 / Details: Microfocus rotating anode with 70 uM focal spot. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Varimax confocal X-ray optical assembly / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. all: 25368 / Num. obs: 23390 / % possible obs: 92.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Biso Wilson estimate: 30.73 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 12.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→17.92 Å / Cor.coef. Fo:Fc: 0.9263 / Cor.coef. Fo:Fc free: 0.9086 / SU R Cruickshank DPI: 0.248 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: Data reduction was accomplished by employing the AutoPROC toolbox and the structure determined by molecular replacement with AMoRE using an apo-state model of S. aureus TMK. COOT was used to ...Details: Data reduction was accomplished by employing the AutoPROC toolbox and the structure determined by molecular replacement with AMoRE using an apo-state model of S. aureus TMK. COOT was used to inspect the model and electron density and BUSTER or REFMAC were used for macromolecular refinement calculations. Geometry of the model was analyzed with MolProbity and stereochemistry of the compound analyzed with MOGUL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.26 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→17.92 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.09 Å / Total num. of bins used: 12
|
