- PDB-4h1w: E1 structure of the (SR) Ca2+-ATPase in complex with Sarcolipin -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4h1w
Title
E1 structure of the (SR) Ca2+-ATPase in complex with Sarcolipin
Components
SERCA1a
Sarcolipin
Keywords
HYDROLASE/HYDROLASE REGULATOR / P-type ATPase / Ca2+ transport / Sarcolipin / Phospholamban / Membrane protein / HYDROLASE-HYDROLASE REGULATOR complex
Function / homology
Function and homology information
positive regulation of protein depolymerization / regulation of ATPase-coupled calcium transmembrane transporter activity / regulation of relaxation of muscle / negative regulation of calcium ion import into sarcoplasmic reticulum / negative regulation of ATPase-coupled calcium transmembrane transporter activity / negative regulation of protein-containing complex disassembly / negative regulation of calcium ion import / positive regulation of calcium ion import into sarcoplasmic reticulum / positive regulation of ATPase-coupled calcium transmembrane transporter activity / positive regulation of fast-twitch skeletal muscle fiber contraction ...positive regulation of protein depolymerization / regulation of ATPase-coupled calcium transmembrane transporter activity / regulation of relaxation of muscle / negative regulation of calcium ion import into sarcoplasmic reticulum / negative regulation of ATPase-coupled calcium transmembrane transporter activity / negative regulation of protein-containing complex disassembly / negative regulation of calcium ion import / positive regulation of calcium ion import into sarcoplasmic reticulum / positive regulation of ATPase-coupled calcium transmembrane transporter activity / positive regulation of fast-twitch skeletal muscle fiber contraction / H zone / calcium ion import into sarcoplasmic reticulum / negative regulation of striated muscle contraction / regulation of striated muscle contraction / sarcoplasmic reticulum calcium ion transport / P-type Ca2+ transporter / P-type calcium transporter activity / positive regulation of cardiac muscle cell contraction / regulation of cardiac muscle contraction by calcium ion signaling / I band / endoplasmic reticulum-Golgi intermediate compartment / regulation of calcium ion transport / sarcoplasmic reticulum membrane / enzyme inhibitor activity / sarcoplasmic reticulum / calcium ion transport / ATPase binding / calcium ion binding / endoplasmic reticulum membrane / perinuclear region of cytoplasm / endoplasmic reticulum / ATP hydrolysis activity / ATP binding / membrane / metal ion binding Similarity search - Function
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj
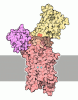


 Assembly
Assembly



 Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
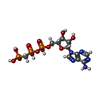
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM
Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Sample preparation
Sample preparation / Beamline: X06SA / Wavelength: 0.97935 Å
/ Beamline: X06SA / Wavelength: 0.97935 Å Processing
Processing