 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4fra: Crystal Structure of ABBA+UDP+Gal at pH 5.0 with MPD as the cryop... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4fra | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of ABBA+UDP+Gal at pH 5.0 with MPD as the cryoprotectant | ||||||
 Components Components | Histo-blood group ABO system transferase | ||||||
 Keywords Keywords | TRANSFERASE / MANGANESE / ABO ROSSMANN FOLD / GLYCOPROTEIN / RETAINING GLYCOSYLTRANSFERASE / BLOOD GROUP ANTIGEN / METAL-BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationfucosylgalactoside 3-alpha-galactosyltransferase / glycoprotein-fucosylgalactoside alpha-N-acetylgalactosaminyltransferase / glycoprotein-fucosylgalactoside alpha-N-acetylgalactosaminyltransferase activity / fucosylgalactoside 3-alpha-galactosyltransferase activity / ABO blood group biosynthesis / hexosyltransferase activity / Golgi cisterna membrane / antigen binding / manganese ion binding / carbohydrate metabolic process ...fucosylgalactoside 3-alpha-galactosyltransferase / glycoprotein-fucosylgalactoside alpha-N-acetylgalactosaminyltransferase / glycoprotein-fucosylgalactoside alpha-N-acetylgalactosaminyltransferase activity / fucosylgalactoside 3-alpha-galactosyltransferase activity / ABO blood group biosynthesis / hexosyltransferase activity / Golgi cisterna membrane / antigen binding / manganese ion binding / carbohydrate metabolic process / vesicle / Golgi membrane / nucleotide binding / Golgi apparatus / extracellular region / membrane / metal ion binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.43 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.43 Å | ||||||
 Authors Authors | Johal, A.R. / Alfaro, J.A. / Blackler, R.J. / Schuman, B. / Borisova, S.N. / Evans, S.V. | ||||||
 Citation Citation | Journal: Glycobiology / Year: 2014 Title: pH-induced conformational changes in human ABO(H) blood group glycosyltransferases confirm the importance of electrostatic interactions in the formation of the semi-closed state. Authors: Johal, A.R. / Blackler, R.J. / Alfaro, J.A. / Schuman, B. / Borisova, S. / Evans, S.V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4fra.cif.gz | 132.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4fra.ent.gz | 102.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4fra.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fr/4fraftp://data.pdbj.org/pub/pdb/validation_reports/fr/4fra | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4fqwC  4frbC  4frdC  4freC  4frhC  4frlC  4frmC  4froC  4frpC  4frqC  4gbpC  4kxoC  1lz7S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 34371.672 Da / Num. of mol.: 1 / Mutation: G176R, A268G Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ABO / Plasmid: PCW DELTA 1AC / Production host:  References: UniProt: H6A2X0, UniProt: P16442*PLUS, fucosylgalactoside 3-alpha-galactosyltransferase |
|---|---|
| #2: Chemical | ChemComp-UDP / 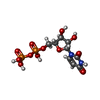  Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM |
| #3: Sugar | ChemComp-GAL / 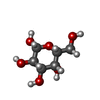  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
| #4: Chemical | ChemComp-PEG /   Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 316 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 316 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.27 Å3/Da / Density % sol: 45.8 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5 Details: 1% PEG 4000, 5% MPD, 5 mM manganese chloride, 100 mM ammonium sulfate, 70 mM sodium chloride,50 mM ADA, 30 mM sodium acetate with 20% MPD as cryoprotectant, pH 5.0, VAPOR DIFFUSION, HANGING ...Details: 1% PEG 4000, 5% MPD, 5 mM manganese chloride, 100 mM ammonium sulfate, 70 mM sodium chloride,50 mM ADA, 30 mM sodium acetate with 20% MPD as cryoprotectant, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-002 / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Nov 26, 2008 / Details: OSMIC BLUE MIRRORS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.43→20 Å / Num. obs: 55128 / % possible obs: 94.8 % / Redundancy: 4.27 % / Rmerge(I) obs: 0.029 / Net I/σ(I): 23.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 29.45 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1LZ7 Resolution: 1.43→20 Å / Cor.coef. Fo:Fc: 0.971 / Cor.coef. Fo:Fc free: 0.963 / Occupancy max: 1 / Occupancy min: 0 / SU B: 0.923 / SU ML: 0.037 / Cross valid method: THROUGHOUT / ESU R: 0.068 / ESU R Free: 0.069 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.995 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.43→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|