 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4crz: Direct visualisation of strain-induced protein prost-translationa... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4crz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Direct visualisation of strain-induced protein prost-translational modification | ||||||
 Components Components |
| ||||||
 Keywords Keywords | LYASE / COENZYME A / RADIATION DAMAGE / PANTOTHENATE | ||||||
| Function / homology |  Function and homology information Function and homology informationalanine biosynthetic process / aspartate 1-decarboxylase / aspartate 1-decarboxylase activity / acetyl-CoA binding / pantothenate biosynthetic process / zymogen activation / acyltransferase activity, transferring groups other than amino-acyl groups / protein autoprocessing / protein processing / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Monteiro, D.C.F. / Patel, V. / Bartlett, C.P. / Grant, T.D. / Nozaki, S. / Gowdy, J.A. / Snell, E.H. / Niki, H. / Pearson, A.R. / Webb, M.E. | ||||||
 Citation Citation | Journal: Chem.Biol. / Year: 2015 Title: The Structure of the Pand/Panz Protein Complex Reveals Negative Feedback Regulation of Pantothenate Biosynthesis by Coenzyme A. Authors: Monteiro, D.C. / Patel, V. / Bartlett, C.P. / Nozaki, S. / Grant, T.D. / Gowdy, J.A. / Thompson, G.S. / Kalverda, A.P. / Snell, E.H. / Niki, H. / Pearson, A.R. / Webb, M.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4crz.cif.gz | 72.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4crz.ent.gz | 52.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4crz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cr/4crzftp://data.pdbj.org/pub/pdb/validation_reports/cr/4crz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4cs0C  4azdS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 15793.928 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Protein | Mass: 15738.804 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
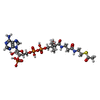
-Non-polymers , 4 types, 148 molecules 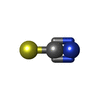


| #3: Chemical | ChemComp-SCN /  Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS |
|---|---|
| #4: Chemical | ChemComp-ACO /  Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S |
| #5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 145 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | HEXAHIS-TAGGED ADC. POINT MUTATION T57V. C-TERMINAL HEXAHIS-TAGGED PANZ. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.4 Details: 20% (W/V) POLYETHYLENE GLYCOL (PEG) 3350, 0.1 M BIS-TRIS PROPANE PH 7.4, 0.2 M POTASSIUM THIOCYANATE |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.542 |
| Detector | Type: RIGAKU R-AXIS IV / Detector: IMAGE PLATE / Date: Dec 3, 2013 / Details: VARIMAX OPTICS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.542 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→33.7 Å / Num. obs: 32244 / % possible obs: 99.7 % / Observed criterion σ(I): 2 / Redundancy: 2.4 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 13.3 |
| Reflection shell | Resolution: 1.7→1.74 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.4 / Mean I/σ(I) obs: 2.2 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4AZD Resolution: 1.7→29.52 Å / Cor.coef. Fo:Fc: 0.977 / Cor.coef. Fo:Fc free: 0.966 / SU B: 1.563 / SU ML: 0.051 / Cross valid method: THROUGHOUT / ESU R: 0.082 / ESU R Free: 0.083 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE BIOLOGICALLY RELEVANT HETEROOCTAMER IS FORMED BY APPLICATION OF THE CRYSTALLOGRAPHIC 4-FOLD SYMMETRY AXIS TO THE ASYMMETRIC UNIT CELL ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE BIOLOGICALLY RELEVANT HETEROOCTAMER IS FORMED BY APPLICATION OF THE CRYSTALLOGRAPHIC 4-FOLD SYMMETRY AXIS TO THE ASYMMETRIC UNIT CELL CONTENTS. EACH ASU CONTAINS ONE ADC PROTOMER AND ONE PANZ PROTOMER.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.383 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→29.52 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|