 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4bz4: CorA is a surface-associated copper-binding protein important in ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4bz4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CorA is a surface-associated copper-binding protein important in Methylomicrobium album BG8 copper acquisition | ||||||
 Components Components | COPPER-REPRESSIBLE POLYPEPTIDE | ||||||
 Keywords Keywords | COPPER-BINDING PROTEIN / COPPER ACQUISITION / METHANOTROPH | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  METHYLOMICROBIUM ALBUM BG8 (bacteria) METHYLOMICROBIUM ALBUM BG8 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.6 Å | ||||||
 Authors Authors | Johnson, K.A. / Ve, T. / Pedersen, R.B. / Lillehaug, J.R. / Jensen, H.B. / Helland, R. / Karlsen, O.A. | ||||||
 Citation Citation | Journal: Plos One / Year: 2014 Title: Cora is a Copper Repressible Surface-Associated Copper(I)-Binding Protein Produced in Methylomicrobium Album Bg8. Authors: Johnson, K.A. / Ve, T. / Larsen, O. / Pedersen, R.B. / Lillehaug, J.R. / Jensen, H.B. / Helland, R. / Karlsen, O.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4bz4.cif.gz | 272.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4bz4.ent.gz | 219.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4bz4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bz/4bz4ftp://data.pdbj.org/pub/pdb/validation_reports/bz/4bz4 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 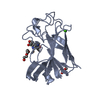
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 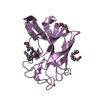
| ||||||||
| 6 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 6 molecules ABCDEF
| #1: Protein | Mass: 25079.221 Da / Num. of mol.: 6 / Source method: isolated from a natural source Details: MODIFICATION OF RESIDUE 62. TRYPTOPHAN IS OXIDIZED TO KYNURENINE Source: (natural) METHYLOMICROBIUM ALBUM BG8 (bacteria) / References: UniProt: P71489, UniProt: H8GPE3*PLUS |
|---|
-Non-polymers , 5 types, 1204 molecules 


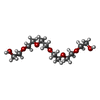

| #2: Chemical | ChemComp-CU1 /  Mass: 63.546 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Cu Mass: 63.546 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Cu#3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-PG4 /  Mass: 194.226 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#5: Chemical | ChemComp-P6G /  Mass: 282.331 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 282.331 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1175 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | THE FIRST RESIDUE VISIBLE IN ELECTRON DENSITY IS NUMBER 28 IN THE SEQUENCE. OTHER RESIDUES NOT ...THE FIRST RESIDUE VISIBLE IN ELECTRON DENSITY IS NUMBER 28 IN THE SEQUENCE. OTHER RESIDUES NOT INCLUDED IN THE STRUCTURE ARE DUE TO LACK OF WELL DEFINED ELECTRON DENSITY |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7 / Details: 11.5-14% PEG 8K, 0.1 BISTRIS PH 7 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9794, 1.5418 / Beamline: ID29 / Wavelength: 0.9794, 1.5418 | |||||||||
| Detector | Type: ADSC CCD / Detector: CCD / Date: Dec 10, 2009 | |||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.6→30 Å / Num. obs: 168264 / % possible obs: 98.6 % / Observed criterion σ(I): 0 / Redundancy: 2.8 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 15.3 | |||||||||
| Reflection shell | Resolution: 1.6→1.64 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.43 / Mean I/σ(I) obs: 2.8 / % possible all: 97 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS Starting model: NONE Resolution: 1.6→30 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.954 / SU B: 1.479 / SU ML: 0.052 / Cross valid method: THROUGHOUT / ESU R: 0.078 / ESU R Free: 0.079 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. DISORDERED REGIONS ARE NOT INCLUDED IN THE STRUCTURE. THE ORIENTATIONS OF MET57 ARE ONLY MODELED, AND GIVEN ZERO OCCUPANCY, DUE TO UNUSUAL ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. DISORDERED REGIONS ARE NOT INCLUDED IN THE STRUCTURE. THE ORIENTATIONS OF MET57 ARE ONLY MODELED, AND GIVEN ZERO OCCUPANCY, DUE TO UNUSUAL ELECTRON DENSITY SURROUNDING THE MAIN CHAIN AND SIDE CHAIN OF THIS AND THE FOLLOWING RESIDUE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.349 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|