 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4atb: Crystal structure of the NF90-NF45 dimerisation domain complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4atb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the NF90-NF45 dimerisation domain complex with CTP | ||||||
 Components Components |
| ||||||
 Keywords Keywords | IMMUNE SYSTEM / TEMPLATE-FREE NUCLEOTIDYL TRANSFERASE FOLD | ||||||
| Function / homology |  Function and homology information Function and homology informationRegulation of CDH11 gene transcription / spliceosome-depend formation of circular RNA / PKR-mediated signaling / mRNA 3'-UTR AU-rich region binding / negative regulation of viral genome replication / Neutrophil degranulation / double-stranded RNA binding / virus receptor activity / defense response to virus / protein phosphorylation ...Regulation of CDH11 gene transcription / spliceosome-depend formation of circular RNA / PKR-mediated signaling / mRNA 3'-UTR AU-rich region binding / negative regulation of viral genome replication / Neutrophil degranulation / double-stranded RNA binding / virus receptor activity / defense response to virus / protein phosphorylation / negative regulation of translation / ribonucleoprotein complex / negative regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / nucleolus / mitochondrion / DNA binding / nucleoplasm / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å | ||||||
 Authors Authors | Wolkowicz, U.M. / Cook, A.G. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2012 Title: NF45 Dimerizes with NF90, Zfr and Spnr Via a Conserved Domain that Has a Nucleotidyltransferase Fold. Authors: Wolkowicz, U.M. / Cook, A.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4atb.cif.gz | 231.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4atb.ent.gz | 182.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4atb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4atb_validation.pdf.gz | 1.1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4atb_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 4atb_validation.xml.gz | 47.8 KB | Display | |
| Data in CIF | 4atb_validation.cif.gz | 65.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/at/4atbftp://data.pdbj.org/pub/pdb/validation_reports/at/4atb | HTTPS FTP |
-Related structure data
| Related structure data |  4at7SC  4at8C  4at9C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.58491, -0.00013, 0.8111), Vector: |
-Components
| #1: Protein | Mass: 40359.840 Da / Num. of mol.: 2 / Fragment: DZF DOMAIN, RESIDUES 29-390 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Protein | Mass: 42526.727 Da / Num. of mol.: 2 / Fragment: DZF DOMAIN, RESIDUES 1-381 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical | 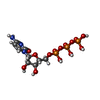  Mass: 483.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H16N3O14P3 Mass: 483.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H16N3O14P3Sequence details | TWO RESIDUES ADDED AT N-TERMINUS DERIVED FROM PRESCISSION CLEAVAGE SITE TWO RESIDUES AT N-TERMINUS ...TWO RESIDUES ADDED AT N-TERMINUS DERIVED FROM PRESCISSIO | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 57 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: 12-14 % PEG3350, 200 MM MGCL2, 100 MM MES PH 6.5 AND 5 % GLYCEROL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 0.9173 / Beamline: I04-1 / Wavelength: 0.9173 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: May 15, 2011 / Details: TOROIDAL MIRROR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9173 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→66 Å / Num. obs: 33184 / % possible obs: 96.8 % / Observed criterion σ(I): 2 / Redundancy: 2.8 % / Biso Wilson estimate: 40.31 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 7.6 |
| Reflection shell | Resolution: 3.1→3.27 Å / Rmerge(I) obs: 0.36 / Mean I/σ(I) obs: 2.8 / % possible all: 98.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4AT7 Resolution: 3.1→66.427 Å / SU ML: 0.38 / σ(F): 1.36 / Phase error: 24.92 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 15.336 Å2 / ksol: 0.325 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.8 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→66.427 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
