| Entry | Database: PDB / ID: 4aff
|
|---|
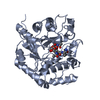
| Title | High resolution structure of a PII mutant (I86N) protein in complex with ATP, MG and FLC |
|---|
 Components Components | NITROGEN REGULATORY PROTEIN P-II |
|---|
 Keywords Keywords | SIGNALING PROTEIN |
|---|
| Function / homology |  Function and homology information Function and homology information
regulation of nitrogen utilization / enzyme regulator activity / ATP binding / identical protein binding / cytosolSimilarity search - Function Nitrogen regulatory protein P-II, urydylation site / P-II protein uridylation site. / Nitrogen regulatory protein PII, conserved site / P-II protein C-terminal region signature. / Nitrogen regulatory protein P-II / P-II protein family profile. / Nitrogen regulatory protein PII / Nitrogen regulatory protein P-II / Alpha-Beta Plaits - #120 / Nitrogen regulatory PII-like, alpha/beta ...Nitrogen regulatory protein P-II, urydylation site / P-II protein uridylation site. / Nitrogen regulatory protein PII, conserved site / P-II protein C-terminal region signature. / Nitrogen regulatory protein P-II / P-II protein family profile. / Nitrogen regulatory protein PII / Nitrogen regulatory protein P-II / Alpha-Beta Plaits - #120 / Nitrogen regulatory PII-like, alpha/beta / Nitrogen regulatory protein PII/ATP phosphoribosyltransferase, C-terminal / Alpha-Beta Plaits / 2-Layer Sandwich / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  SYNECHOCOCCUS ELONGATUS (bacteria) SYNECHOCOCCUS ELONGATUS (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.05 Å |
|---|
 Authors Authors | Zeth, K. / Fokina, O. / Chellamuthu, V.R. / Forchhammer, K. |
|---|
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2012
Title: An Engineered Pii Protein Variant that Senses a Novel Ligand: Atomic Resolution Structure of the Complex with Citrate.
Authors: Zeth, K. / Fokina, O. / Forchhammer, K. |
|---|
| History | | Deposition | Jan 19, 2012 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Feb 1, 2012 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 25, 2012 | Group: Database references / Structure summary |
|---|
| Revision 1.2 | Aug 15, 2012 | Group: Database references |
|---|
| Revision 1.3 | May 8, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj





 Assembly
Assembly


 Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM
Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM
 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg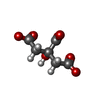
 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7
Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X10SA / Wavelength: 1
/ Beamline: X10SA / Wavelength: 1  Processing
Processing