 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 459d | ||||||
|---|---|---|---|---|---|---|---|
| Title | DNA MINOR-GROOVE RECOGNITION OF A TRIS-BENZIMIDAZOLE DRUG | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA / MINOR GROOVE BINDING / B-DNA / COMPLEXED WITH TRIS-BENZIMIDAZOLE / DEOXYRIBONUCLEIC ACID | ||||||
| Function / homology | Chem-TBZ / DNA / DNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.3 Å X-RAY DIFFRACTION / Resolution: 2.3 Å | ||||||
 Authors Authors | Aymami, J. / Nunn, C.M. / Neidle, S. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 1999 Title: DNA minor groove recognition of a non-self-complementary AT-rich sequence by a tris-benzimidazole ligand. Authors: Aymami, J. / Nunn, C.M. / Neidle, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 459d.cif.gz | 27.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb459d.ent.gz | 17.4 KB | Display | PDB format |
| PDBx/mmJSON format | 459d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 459d_validation.pdf.gz | 591.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 459d_full_validation.pdf.gz | 594.9 KB | Display | |
| Data in XML | 459d_validation.xml.gz | 5.8 KB | Display | |
| Data in CIF | 459d_validation.cif.gz | 7.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/59/459dftp://data.pdbj.org/pub/pdb/validation_reports/59/459d | HTTPS FTP |
-Related structure data






-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 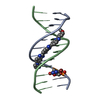
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3732.286 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: COMPLEXED WITH 2''-(4-METHOXYPHENYL)-5-(3-AMINO-1-PYRROLIDINYL)-2,5',2', 5''- TRI-BENZIMIDAZOLE |
|---|---|
| #2: DNA chain | Mass: 3671.418 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: COMPLEXED WITH 2''-(4-METHOXYPHENYL)-5-(3-AMINO-1-PYRROLIDINYL)-2,5',2', 5''- TRI-BENZIMIDAZOLE |
| #3: Chemical | ChemComp-TBZ / 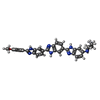  Mass: 541.626 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H29N8O Mass: 541.626 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H29N8O |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 112 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 112 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.56 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 13 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 288 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.2 Å / Num. obs: 3281 / % possible obs: 87.7 % / Observed criterion σ(I): 0 / Rmerge(I) obs: 0.05 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.3→8 Å / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.285 / Rfactor Rfree: 0.361 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
