| Entry | Database: PDB / ID: 3wvd
|
|---|
| Title | Crystal structure of Nitrile Hydratase mutant bR56K complexed with Trimethylacetonitrile, photo-activated for 50 min |
|---|
 Components Components | (Nitrile hydratase subunit ...) x 2 |
|---|
 Keywords Keywords | LYASE / cysteine sulfinic acid / Cys-SO2H / Cys-SOH |
|---|
| Function / homology |  Function and homology information Function and homology information
Nitrile Hydratase; Chain A / Nitrile hydratase alpha /Thiocyanate hydrolase gamma / Nitrile hydratase, beta subunit / Nitrile hydratase, alpha subunit / Nitrile hydratase, beta subunit / Nitrile hydratase beta subunit domain / Nitrile hydratase beta subunit, N-terminal / : / Nitrile hydratase beta subunit, C-terminal / Nitrile hydratase beta subunit, N-terminal ...Nitrile Hydratase; Chain A / Nitrile hydratase alpha /Thiocyanate hydrolase gamma / Nitrile hydratase, beta subunit / Nitrile hydratase, alpha subunit / Nitrile hydratase, beta subunit / Nitrile hydratase beta subunit domain / Nitrile hydratase beta subunit, N-terminal / : / Nitrile hydratase beta subunit, C-terminal / Nitrile hydratase beta subunit, N-terminal / Nitrile hydratase alpha subunit /Thiocyanate hydrolase gamma subunit / Nitrile hydratase alpha /Thiocyanate hydrolase gamma / Nitrile hydratase, alpha chain / Nitrile hydratase alpha /Thiocyanate hydrolase gamma superfamily / SH3 type barrels. - #50 / Electron transport accessory-like domain superfamily / Cyclin A; domain 1 / SH3 type barrels. / Roll / Alpha-Beta Complex / Orthogonal Bundle / Mainly Beta / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology : / 2,2-dimethylpropanenitrile / Iron-containing nitrile hydratase subunit alpha / Iron-containing nitrile hydratase subunit betaSimilarity search - Component |
|---|
| Biological species |  Rhodococcus erythropolis (bacteria) Rhodococcus erythropolis (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.18 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.18 Å |
|---|
 Authors Authors | Yamanaka, Y. / Hashimoto, K. / Noguchi, K. / Yohda, M. / Odaka, M. |
|---|
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2015
Title: Time-Resolved Crystallography of the Reaction Intermediate of Nitrile Hydratase: Revealing a Role for the Cysteinesulfenic Acid Ligand as a Catalytic Nucleophile.
Authors: Yamanaka, Y. / Kato, Y. / Hashimoto, K. / Iida, K. / Nagasawa, K. / Nakayama, H. / Dohmae, N. / Noguchi, K. / Noguchi, T. / Yohda, M. / Odaka, M. |
|---|
| History | | Deposition | May 17, 2014 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Jun 17, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 4, 2015 | Group: Database references |
|---|
| Revision 1.2 | Oct 30, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Structure summary
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature / pdbx_struct_conn_angle / struct_conn / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_asym_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj


 Assembly
Assembly


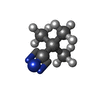

 Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe
Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 83.132 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9N
Mass: 83.132 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9N Sample preparation
Sample preparation / Beamline: BL-1A / Wavelength: 1 Å
/ Beamline: BL-1A / Wavelength: 1 Å Processing
Processing