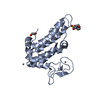
- PDB-3uwt: Crystal structure of a RNA binding domain of poly-U binding splic... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 3uwt
タイトル
Crystal structure of a RNA binding domain of poly-U binding splicing factor 60KDa (PUF60) from Homo sapiens at 2.50 A resolution
要素
Poly(U)-binding-splicing factor PUF60
キーワード
RNA BINDING PROTEIN / RNA recognition motive / RRM / RNA binding domain / splicing / Structural Genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-BIOLOGY / Partnership for T-Cell Biology / TCELL
機能・相同性
機能・相同性情報
mRNA splice site recognition / alternative mRNA splicing, via spliceosome / regulation of alternative mRNA splicing, via spliceosome / mRNA Splicing - Major Pathway / cell junction / cadherin binding / ribonucleoprotein complex / apoptotic process / DNA binding / RNA binding ...mRNA splice site recognition / alternative mRNA splicing, via spliceosome / regulation of alternative mRNA splicing, via spliceosome / mRNA Splicing - Major Pathway / cell junction / cadherin binding / ribonucleoprotein complex / apoptotic process / DNA binding / RNA binding / nucleoplasm / identical protein binding 類似検索 - 分子機能
THIS CONSTRUCT (RESIDUES 118-316) WAS EXPRESSED WITH A PURIFICATION TAG MGSDKIHHHHHHENLYFQG. THE ...THIS CONSTRUCT (RESIDUES 118-316) WAS EXPRESSED WITH A PURIFICATION TAG MGSDKIHHHHHHENLYFQG. THE TAG WAS REMOVED WITH TEV PROTEASE LEAVING ONLY A GLYCINE (0) FOLLOWED BY THE TARGET SEQUENCE. RESIDUE NUMBERING IS BASED ON ISOFORM 1 OF UNIPROTKB Q9UHX1.
解像度: 2.5→29.798 Å / Num. all: 6937 / Num. obs: 6937 / % possible obs: 99.8 % / 冗長度: 6.5 % / Biso Wilson estimate: 68.061 Å2 / Rsym value: 0.094 / Net I/σ(I): 11.4
反射 シェル
Diffraction-ID: 1
解像度 (Å)
冗長度 (%)
Mean I/σ(I) obs
Num. measured all
Num. unique all
Rsym value
% possible all
Rmerge(I) obs
2.5-2.57
6.9
1.6
3532
514
1.377
99.9
2.57-2.64
6.8
0.7
3254
478
1.003
99.6
0.014
2.64-2.71
6.7
1
3205
475
0.724
99.7
0.014
2.71-2.8
6.8
1.3
3200
473
0.595
99.8
0.014
2.8-2.89
6.8
1.9
2982
439
0.406
99.8
0.014
2.89-2.99
6.7
2.5
2871
430
0.304
99.8
0.014
2.99-3.1
6.8
3.2
2887
426
0.237
100
0.014
3.1-3.23
6.7
4.6
2691
402
0.163
99.8
0.014
3.23-3.37
6.6
5.8
2613
395
0.13
99.8
0.014
3.37-3.54
6.6
7.4
2502
377
0.098
99.8
0.014
3.54-3.73
6.3
5.8
2221
353
0.118
99.9
0.014
3.73-3.95
6.4
9.6
2222
348
0.071
99.9
0.014
3.95-4.23
6.4
11
2015
314
0.061
100
0.014
4.23-4.56
6.4
12.9
1919
301
0.05
99.9
0.014
4.56-5
6.1
12
1728
281
0.051
99.8
0.014
5-5.59
5.9
10.6
1492
251
0.055
99.9
0.014
5.59-6.46
5.6
9.1
1310
234
0.071
99.7
0.014
6.46-7.91
6.1
8.8
1209
197
0.07
99.9
0.014
7.91-11.18
6.2
12.1
970
157
0.052
99.7
0.014
11.18-29.798
5.5
13.9
508
92
0.045
94.7
0.014
-
位相決定
位相決定
手法: 多波長異常分散
-
解析
ソフトウェア
名称
バージョン
分類
NB
MolProbity
3beta29
モデル構築
PDB_EXTRACT
3.1
データ抽出
SHELX
位相決定
SHARP
位相決定
SCALA
3.3.20
データスケーリング
BUSTER-TNT
2.10.0
精密化
MOSFLM
データ削減
SHELXD
位相決定
BUSTER
2.10.0
精密化
精密化
構造決定の手法: 多波長異常分散 / 解像度: 2.5→29.798 Å / Cor.coef. Fo:Fc: 0.9446 / Cor.coef. Fo:Fc free: 0.9143 / Occupancy max: 1 / Occupancy min: 0.5 / 交差検証法: THROUGHOUT / σ(F): 0 詳細: 1. A MET-INHIBITION PROTOCOL WAS USED FOR SELENOMETHIONINE INCORPORATION DURING PROTEIN EXPRESSION. THE OCCUPANCY OF THE SE ATOMS IN THE MSE RESIDUES WAS REDUCED TO 0.75 FOR THE REDUCED ...詳細: 1. A MET-INHIBITION PROTOCOL WAS USED FOR SELENOMETHIONINE INCORPORATION DURING PROTEIN EXPRESSION. THE OCCUPANCY OF THE SE ATOMS IN THE MSE RESIDUES WAS REDUCED TO 0.75 FOR THE REDUCED SCATTERING POWER DUE TO PARTIAL S-MET INCORPORATION. 2. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS. 3. CHLORIDE (CL) FROM THE CRYSTALLIZATION SOLUTION HAS BEEN MODELED IN THE SOLVENT STRUCTURE. 4. THE REFINEMENT WAS RESTRAINED WITH THE MAD PHASES. 5. RAMACHANDRAN OUTLIERS AT RESIDUES 150 AND 290 ARE SUPPORTED BY ELECTRON DENSITY.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード








 PDBj
PDBj


 集合体
集合体


 分子量: 35.453 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Cl
分子量: 35.453 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Cl 分子量: 18.015 Da / 分子数: 27 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 27 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 8.2.2 / 波長: 0.9537,0.9796,0.9793
/ ビームライン: 8.2.2 / 波長: 0.9537,0.9796,0.9793 解析
解析