 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3rty: Structure of an Enclosed Dimer Formed by The Drosophila Period Protein -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rty | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of an Enclosed Dimer Formed by The Drosophila Period Protein | ||||||
 Components Components | Period circadian protein | ||||||
 Keywords Keywords | CIRCADIAN CLOCK PROTEIN / PAS domain / signalling / Timeless | ||||||
| Function / homology |  Function and homology information Function and homology informationNuclear import of PER and TIM / Dephosphorylation of TIM / eclosion rhythm / Transcription repression by PER and activation by PDP1 / Dephosphorylation of PER / Phosphorylation of PER and TIM / copulation / Degradation of PER / Degradation of TIM / male courtship behavior, veined wing generated song production ...Nuclear import of PER and TIM / Dephosphorylation of TIM / eclosion rhythm / Transcription repression by PER and activation by PDP1 / Dephosphorylation of PER / Phosphorylation of PER and TIM / copulation / Degradation of PER / Degradation of TIM / male courtship behavior, veined wing generated song production / circadian temperature homeostasis / circadian sleep/wake cycle / courtship behavior / rhythmic behavior / regulation of locomotor rhythm / regulation of circadian sleep/wake cycle, sleep / entrainment of circadian clock / circadian behavior / mating behavior / response to temperature stimulus / entrainment of circadian clock by photoperiod / locomotor rhythm / response to light stimulus / long-term memory / behavioral response to cocaine / transcription corepressor binding / determination of adult lifespan / transcription coregulator activity / circadian regulation of gene expression / circadian rhythm / regulation of circadian rhythm / transcription corepressor activity / cell body / response to oxidative stress / transcription cis-regulatory region binding / negative regulation of DNA-templated transcription / perinuclear region of cytoplasm / negative regulation of transcription by RNA polymerase II / nucleoplasm / identical protein binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.85 Å | ||||||
 Authors Authors | King, H.A. / Hoelz, A. / Crane, B.R. / Young, M.W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2011 Title: Structure of an enclosed dimer formed by the Drosophila period protein. Authors: King, H.A. / Hoelz, A. / Crane, B.R. / Young, M.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rty.cif.gz | 492 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rty.ent.gz | 403.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3rty.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rt/3rtyftp://data.pdbj.org/pub/pdb/validation_reports/rt/3rty | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 38186.922 Da / Num. of mol.: 8 / Fragment: central fragment (UNP residues 236-574) Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-DTT / 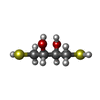  Mass: 154.251 Da / Num. of mol.: 28 / Source method: obtained synthetically / Formula: C4H10O2S2 Mass: 154.251 Da / Num. of mol.: 28 / Source method: obtained synthetically / Formula: C4H10O2S2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.4 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 9 Details: 200 mM lithium chloride, 20 % (w/v) PEG 3350, 100 mM Bicine, pH 9.0, VAPOR DIFFUSION, HANGING DROP, temperature 294K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.1 / Wavelength: 1 Å / Beamline: 8.2.1 / Wavelength: 1 Å |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.85→30 Å / Num. obs: 62952 / % possible obs: 92.4 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 3.3 % / Biso Wilson estimate: 87.3 Å2 / Rsym value: 0.081 / Net I/σ(I): 14.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.85→20.01 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 74354.63 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 26.1009 Å2 / ksol: 0.28 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 70.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.85→20.01 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.85→3.03 Å / Rfactor Rfree error: 0.013 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
