 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rml | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human Thrombin in complex with MI331 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Serine Protease / Kringle / Hydrolase / Blood Coagulation / Blood Clotting / Convertion of Fibrinogen to Fibrin / Hirudin / Blood / glycosylation / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / thrombospondin receptor activity / thrombin / thrombin-activated receptor signaling pathway / Defective factor XII causes hereditary angioedema / negative regulation of astrocyte differentiation / regulation of blood coagulation / neutrophil-mediated killing of gram-negative bacterium / positive regulation of phospholipase C-activating G protein-coupled receptor signaling pathway / Defective F8 cleavage by thrombin ...: / thrombospondin receptor activity / thrombin / thrombin-activated receptor signaling pathway / Defective factor XII causes hereditary angioedema / negative regulation of astrocyte differentiation / regulation of blood coagulation / neutrophil-mediated killing of gram-negative bacterium / positive regulation of phospholipase C-activating G protein-coupled receptor signaling pathway / Defective F8 cleavage by thrombin / Platelet Aggregation (Plug Formation) / positive regulation of collagen biosynthetic process / negative regulation of platelet activation / negative regulation of blood coagulation / negative regulation of fibrinolysis / positive regulation of blood coagulation / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / : / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins / fibrinolysis / regulation of cytosolic calcium ion concentration / : / negative regulation of proteolysis / negative regulation of cytokine production involved in inflammatory response / Regulation of Complement cascade / positive regulation of release of sequestered calcium ion into cytosol / acute-phase response / Cell surface interactions at the vascular wall / Peptide ligand-binding receptors / positive regulation of receptor signaling pathway via JAK-STAT / growth factor activity / serine-type endopeptidase inhibitor activity / lipopolysaccharide binding / platelet activation / response to wounding / positive regulation of protein localization to nucleus / Golgi lumen / positive regulation of reactive oxygen species metabolic process / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / blood coagulation / heparin binding / Thrombin signalling through proteinase activated receptors (PARs) / Dengue Virus-Host Interactions / antimicrobial humoral immune response mediated by antimicrobial peptide / extracellular matrix / blood microparticle / G alpha (q) signalling events / cell surface receptor signaling pathway / endoplasmic reticulum lumen / receptor ligand activity / serine-type endopeptidase activity / signaling receptor binding / calcium ion binding / proteolysis / : / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Hirudo medicinalis (medicinal leech) Hirudo medicinalis (medicinal leech) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.53 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.53 Å | ||||||
 Authors Authors | Biela, A. / Heine, A. / Klebe, G. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2012 Title: Ligand binding stepwise disrupts water network in thrombin: enthalpic and entropic changes reveal classical hydrophobic effect Authors: Biela, A. / Sielaff, F. / Terwesten, F. / Heine, A. / Steinmetzer, T. / Klebe, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rml.cif.gz | 145.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rml.ent.gz | 112.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3rml.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rm/3rmlftp://data.pdbj.org/pub/pdb/validation_reports/rm/3rml | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3rlwC  3rlyC  3rm0C  3rm2C  3rmmC  3rmnC  3rmoC  3t5fC  3uwjC  1h8dS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |






















-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein/peptide , 2 types, 2 molecules LI
| #1: Protein/peptide | Mass: 4096.534 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00734, thrombin |
|---|---|
| #3: Protein/peptide | Mass: 1662.683 Da / Num. of mol.: 1 / Fragment: residues in UNP 60-72 / Source method: obtained synthetically Details: Synthetic Fragment of Hirudin from Hirudo Medicinalis Source: (synth.) Hirudo medicinalis (medicinal leech) / References: UniProt: P09945 |
-Protein / Sugars , 2 types, 2 molecules H
| #2: Protein | Mass: 29780.219 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00734, thrombin |
|---|---|
| #4: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
-Non-polymers , 5 types, 303 molecules 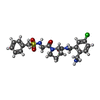




| #5: Chemical | ChemComp-M31 /  Type: peptide-like, Peptide-like / Class: Thrombin inhibitor / Mass: 478.992 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H27ClN4O4S Type: peptide-like, Peptide-like / Class: Thrombin inhibitor / Mass: 478.992 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H27ClN4O4SReferences: N-(BENZYLSULFONYL)GLYCYL-N-[2-(AMINOMETHYL)-5-CHLOROBENZYL]-L-PROLINAMIDE | ||||||
|---|---|---|---|---|---|---|---|
| #6: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#7: Chemical | ChemComp-PO4 / |  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#8: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 297 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.31 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 15% PEG 8000, 20mM sodium phosphate, 175mM sodium chloride, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 277.15K |
-Data collection
| Diffraction | Mean temperature: 108 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06DA / Wavelength: 1 Å / Beamline: X06DA / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jun 22, 2009 / Details: Collimating mirror |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.53→50 Å / Num. all: 50478 / Num. obs: 50478 / % possible obs: 96.4 % / Redundancy: 2.9 % / Biso Wilson estimate: 14.43 Å2 / Rsym value: 0.035 / Net I/σ(I): 29.2 |
| Reflection shell | Resolution: 1.53→1.56 Å / Redundancy: 2.7 % / Mean I/σ(I) obs: 2.7 / Num. unique all: 2436 / Rsym value: 0.357 / % possible all: 92.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1H8D Resolution: 1.53→35.661 Å / Occupancy max: 1 / Occupancy min: 0.28 / FOM work R set: 0.8904 / SU ML: 0.17 / Cross valid method: Free R / σ(F): 0 / Phase error: 18.15 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 47.984 Å2 / ksol: 0.389 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 91.76 Å2 / Biso mean: 24.4019 Å2 / Biso min: 7.79 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.53→35.661 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
