 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3qz1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Bovine Steroid of 21-hydroxylase (P450c21) | ||||||
 Components Components | Steroid 21-hydroxylase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / P450 monooxygenase / 21-hydroxylase | ||||||
| Function / homology |  Function and homology information Function and homology informationGlucocorticoid biosynthesis / Mineralocorticoid biosynthesis / steroid 21-monooxygenase / steroid 21-monooxygenase activity / 17-hydroxyprogesterone 21-hydroxylase activity / progesterone 21-hydroxylase activity / Endogenous sterols / glucocorticoid biosynthetic process / steroid biosynthetic process / steroid hydroxylase activity ...Glucocorticoid biosynthesis / Mineralocorticoid biosynthesis / steroid 21-monooxygenase / steroid 21-monooxygenase activity / 17-hydroxyprogesterone 21-hydroxylase activity / progesterone 21-hydroxylase activity / Endogenous sterols / glucocorticoid biosynthetic process / steroid biosynthetic process / steroid hydroxylase activity / steroid metabolic process / steroid binding / iron ion binding / heme binding / endoplasmic reticulum membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3 Å | ||||||
 Authors Authors | Zhao, B. / Waterman, M.R. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of Bovine Steroid of 21-hydroxylase (P450c21) Authors: Zhao, B. / Lei, L. / Sundaramoorthy, M. / Kagawa, N. / Waterman, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3qz1.cif.gz | 371.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3qz1.ent.gz | 303.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3qz1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qz/3qz1ftp://data.pdbj.org/pub/pdb/validation_reports/qz/3qz1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 56160.270 Da / Num. of mol.: 4 / Mutation: T241R, L442A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4#3: Chemical | ChemComp-3QZ / ( 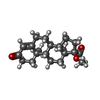  Mass: 330.461 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C21H30O3 / Comment: hormone*YM Mass: 330.461 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C21H30O3 / Comment: hormone*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | THE LIGAND 3QZ IN THE STRUCTURE IS 17-HYDROXYPROGESTERONE, WHICH IS VERY HYDROPHOBIC AND ITS ...THE LIGAND 3QZ IN THE STRUCTURE IS 17-HYDROXYPRO | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 56.57 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: PEG 3350, Tacsimate, pH 7.4, VAPOR DIFFUSION, HANGING DROP, temperature 295K |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection twin | Operator: -h,k,-l / Fraction: 0.5 | ||||||||||||||||||
| Reflection | Resolution: 3→50 Å / Num. all: 49955 / Num. obs: 47687 / % possible obs: 95 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Rmerge(I) obs: 0.092 / Rsym value: 0.065 / Net I/σ(I): 4.5 | ||||||||||||||||||
| Reflection shell | Resolution: 3→3.03 Å / Redundancy: 3.2 % / Rmerge(I) obs: 0.539 / Mean I/σ(I) obs: 1.21 / Rsym value: 0.672 / % possible all: 91.9 |

- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3→30 Å / Occupancy max: 1 / Occupancy min: 0.5 / σ(F): 1541
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 139.685 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 323.81 Å2 / Biso mean: 119.1571 Å2 / Biso min: 51.25 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.54 Å / Luzzati d res low obs: 5 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
|
