 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3m1h: Crystal Structure Analysis of the K3 Cleaved Adhesin Domain of Ly... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3m1h | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure Analysis of the K3 Cleaved Adhesin Domain of Lys-gingipain (Kgp) from Porphyromonas gingivalis w83 | ||||||
 Components Components | Lysine specific cysteine protease | ||||||
 Keywords Keywords | CELL INVASION / beta jelly roll barrel / cleaved adhesin family / lys-gingipain / hemagglutination domain / carbohydrate binding / Protease | ||||||
| Function / homology |  Function and homology information Function and homology informationgingipain K / hemolysis in another organism / cysteine-type endopeptidase activity / calcium ion binding / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  Porphyromonas gingivalis (bacteria) Porphyromonas gingivalis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.56 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.56 Å | ||||||
 Authors Authors | Li, N. / Collyer, C.A. / Hunter, N. | ||||||
 Citation Citation | Journal: Mol Microbiol / Year: 2011 Title: The modular structure of haemagglutinin/adhesin regions in gingipains of Porphyromonas gingivalis. Authors: Nan Li / Peter Yun / Cy M Jeffries / David Langley / Roland Gamsjaeger / W Bret Church / Neil Hunter / Charles A Collyer /  Abstract: High-molecular-weight arginine- and lysine-specific (Kgp) gingipains are essential virulence factors expressed by the oral pathogen Porphyromonas gingivalis. Haemagglutinin/adhesin (HA) regions of ...High-molecular-weight arginine- and lysine-specific (Kgp) gingipains are essential virulence factors expressed by the oral pathogen Porphyromonas gingivalis. Haemagglutinin/adhesin (HA) regions of these proteases have been implicated in targeting catalytic domains to biological substrates and in other adhesive functions. We now report the crystal structure of the K3 adhesin domain/module of Kgp, which folds into the distinct β-jelly roll sandwich topology previously observed for K2. A conserved structural feature of K3, previously observed in the Kgp K2 module, is the half-way point anchoring of the surface exposed loops via an arginine residue found in otherwise highly variable sequences. Small-angle X-ray scattering data for the recombinant construct K1K2K3 confirmed a structure comprising a tandem repeat of three homologous modules, K1, K2 and K3 while also indicating an unusual 'y'-shape arrangement of the modules connected by variable linker sequences. Only the K2 and K3 modules and a K1K2 construct were observed to be potently haemolytic. K2, K3 and the K1K2 construct showed preferential recognition of haem-albumin over albumin whereas only low affinity binding was detected for K1 and the K1K2K3 construct. The data indicate replication of some biological functions over the three adhesin domains of Kgp while other functions are restricted. #1: Journal: Eur J Microbiol Immunol / Year: 2011Title: Gingipains from Porphyromonas gingivalis complex domain structures confer diverse functions. Authors: Li, N. / Collyer, C.A. #2: Journal: Eur J Microbiol Immunol / Year: 2013Title: The lysine gingipain adhesin domains from porphyromonas gingivalis interact with erythrocytes and albumin: Structures correlate to function. Authors: Li, N. / Collyer, C.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3m1h.cif.gz | 169 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3m1h.ent.gz | 129.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3m1h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m1/3m1hftp://data.pdbj.org/pub/pdb/validation_reports/m1/3m1h | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 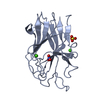 3km5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



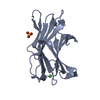








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 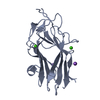
| ||||||||
| 2 | 
| ||||||||
| 3 | 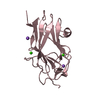
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18978.600 Da / Num. of mol.: 4 / Fragment: K3 cleaved adhesin domain, residues 1427-1602 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Porphyromonas gingivalis (bacteria) / Strain: W83 / Gene: kgp / Plasmid: pET32 / Production host: #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Na#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 825 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 825 / Source method: isolated from a natural source / Formula: H2OSequence details | SEQUENCE DISCREPANC | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 43.98 % / Mosaicity: 0.471 ° |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 0.2M Calcium acetate, 30% PEG 8000, 0.1M Na cacodylate, pH 6.5, vapor diffusion, sitting drop, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron / Beamline: MX1 / Wavelength: 0.95663 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Jan 30, 2009 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.95663 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.56→50 Å / Num. obs: 89782 / % possible obs: 96.7 % / Redundancy: 2.4 % / Biso Wilson estimate: 15.1 Å2 / Rmerge(I) obs: 0.032 / Χ2: 1 / Net I/σ(I): 19.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3KM5 Resolution: 1.56→23.24 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.954 / WRfactor Rfree: 0.203 / WRfactor Rwork: 0.169 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.896 / SU B: 1.379 / SU ML: 0.05 / SU R Cruickshank DPI: 0.08 / SU Rfree: 0.082 / Cross valid method: THROUGHOUT / ESU R: 0.08 / ESU R Free: 0.082 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 52.21 Å2 / Biso mean: 18.413 Å2 / Biso min: 6.98 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.56→23.24 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.563→1.604 Å / Total num. of bins used: 20
|
