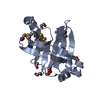
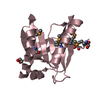
| Entry | Database: PDB / ID: 3lur
|
|---|
| Title | Crystal structure of Putative bacterial transcription regulation protein (NP_372959.1) from Staphylococcus aureus MU50 at 1.81 A resolution |
|---|
 Components Components | Putative bacterial transcription regulation protein |
|---|
 Keywords Keywords | Transcription activator / Structural Genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-2 / Bacterial transcription activator / effector binding domain |
|---|
| Function / homology |  Function and homology information Function and homology information
: / Integron-associated effector binding protein / Integron-associated effector binding protein / Bacterial transcription activator, effector binding / Bacterial transcription activator, effector binding domain / GyrI-like small molecule binding domain / GyrI-like small molecule binding domain / Multidrug-efflux Transporter 1 Regulator Bmrr; Chain A / Regulatory factor, effector binding domain / Regulatory factor, effector binding domain superfamily ...: / Integron-associated effector binding protein / Integron-associated effector binding protein / Bacterial transcription activator, effector binding / Bacterial transcription activator, effector binding domain / GyrI-like small molecule binding domain / GyrI-like small molecule binding domain / Multidrug-efflux Transporter 1 Regulator Bmrr; Chain A / Regulatory factor, effector binding domain / Regulatory factor, effector binding domain superfamily / Alpha-Beta Barrel / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.81 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.81 Å |
|---|
 Authors Authors | Joint Center for Structural Genomics (JCSG) |
|---|
 Citation Citation | Journal: To be published
Title: Crystal structure of Putative bacterial transcription regulation protein (NP_372959.1) from Staphylococcus aureus MU50 at 1.81 A resolution
Authors: Joint Center for Structural Genomics (JCSG) |
|---|
| History | | Deposition | Feb 18, 2010 | Deposition site: RCSB / Processing site: RCSB |
|---|
| Revision 1.0 | Mar 9, 2010 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 13, 2011 | Group: Advisory / Version format compliance |
|---|
| Revision 1.2 | Nov 8, 2017 | Group: Refinement description / Category: software / Item: _software.classification / _software.name |
|---|
| Revision 1.3 | Jul 17, 2019 | Group: Data collection / Derived calculations / Refinement description
Category: software / struct_conn
Item: _software.classification / _software.contact_author ..._software.classification / _software.contact_author / _software.contact_author_email / _software.language / _software.location / _software.name / _software.type / _software.version / _struct_conn.pdbx_leaving_atom_flag |
|---|
| Revision 1.4 | Feb 1, 2023 | Group: Database references / Derived calculations / Category: database_2 / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
| Revision 1.5 | Oct 30, 2024 | Group: Data collection / Refinement description / Structure summary
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / pdbx_entry_details / pdbx_modification_feature / struct_ncs_dom_lim
Item: _pdbx_entry_details.has_protein_modification / _struct_ncs_dom_lim.beg_auth_comp_id ..._pdbx_entry_details.has_protein_modification / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj Assembly
Assembly