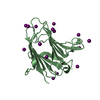
protein transport by the Sec complex / protein secretion by the type II secretion system / 加水分解酵素; プロテアーゼ; ペプチド結合加水分解酵素; その他のペプチターゼ / peptidoglycan catabolic process / metalloendopeptidase activity / endopeptidase activity / proteolysis / extracellular space / metal ion binding 類似検索 - 分子機能
冗長度: 3.5 % / Av σ(I) over netI: 23.39 / 数: 109259 / Rmerge(I) obs: 0.095 / Χ2: 1.06 / D res high: 2.14 Å / D res low: 50 Å / Num. obs: 31654 / % possible obs: 99.7
Diffraction reflection shell
最高解像度 (Å)
最低解像度 (Å)
% possible obs (%)
ID
Rmerge(I) obs
Chi squared
Redundancy
4.61
50
100
1
0.045
1.073
3.8
3.66
4.61
100
1
0.047
1.063
3.7
3.2
3.66
100
1
0.06
0.999
3.7
2.9
3.2
100
1
0.088
0.977
3.7
2.7
2.9
100
1
0.13
0.984
3.6
2.54
2.7
100
1
0.186
1.017
3.6
2.41
2.54
100
1
0.229
1.066
3.5
2.31
2.41
100
1
0.261
1.129
3.3
2.22
2.31
99.5
1
0.278
1.179
3
2.14
2.22
98
1
0.299
1.21
2.6
反射
解像度: 2.14→50 Å / Num. obs: 32280 / % possible obs: 100 % / 冗長度: 3.7 % / Rmerge(I) obs: 0.054 / Χ2: 1.027 / Net I/σ(I): 14.7
反射 シェル
解像度 (Å)
冗長度 (%)
Rmerge(I) obs
Num. unique all
Χ2
Diffraction-ID
% possible all
2.14-2.22
3.6
0.1
3264
1.063
1
100
2.22-2.31
3.7
0.095
3216
1.088
1
100
2.31-2.41
3.7
0.086
3203
1.04
1
100
2.41-2.54
3.7
0.079
3261
1.065
1
100
2.54-2.7
3.7
0.071
3217
1.031
1
100
2.7-2.9
3.7
0.06
3209
0.992
1
100
2.9-3.2
3.7
0.05
3222
0.994
1
100
3.2-3.66
3.7
0.042
3216
1.038
1
100
3.66-4.61
3.7
0.037
3231
0.965
1
100
4.61-50
3.7
0.037
3241
1
1
99.9
-
位相決定
位相決定
手法: 多波長異常分散
-
解析
ソフトウェア
名称
バージョン
分類
NB
DENZO
データ削減
SCALEPACK
データスケーリング
SOLVE
位相決定
RESOLVE
位相決定
REFMAC
精密化
PDB_EXTRACT
3.005
データ抽出
精密化
構造決定の手法: 多波長異常分散 / 解像度: 2.14→37.57 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.919 / WRfactor Rfree: 0.189 / WRfactor Rwork: 0.145 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.891 / SU B: 3.684 / SU ML: 0.098 / SU R Cruickshank DPI: 0.322 / SU Rfree: 0.189 / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.322 / ESU R Free: 0.189 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES: REFINED INDIVIDUALLY
Rfactor
反射数
%反射
Selection details
Rfree
0.195
882
5.1 %
RANDOM
Rwork
0.149
-
-
-
obs
0.151
17436
99.98 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Pseudomonas aeruginosa (緑膿菌)
Pseudomonas aeruginosa (緑膿菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体



 分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn
分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn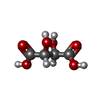
 分子量: 150.087 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H6O6
分子量: 150.087 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H6O6
 分子量: 92.094 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C3H8O3
分子量: 92.094 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C3H8O3 分子量: 18.015 Da / 分子数: 391 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 391 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 19-ID / 波長: 1.28402, 1.28348, 1.2574
/ ビームライン: 19-ID / 波長: 1.28402, 1.28348, 1.2574 解析
解析