 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3guj: T4 lysozyme M102E/L99A mutant with buried charge in apolar cavity... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3guj | ||||||
|---|---|---|---|---|---|---|---|
| Title | T4 lysozyme M102E/L99A mutant with buried charge in apolar cavity--Benzene binding | ||||||
 Components Components | Lysozyme | ||||||
 Keywords Keywords | HYDROLASE / T4 lysozyme / apolar cavity / buried charge / ligand binding / Antimicrobial / Bacteriolytic enzyme / Glycosidase | ||||||
| Function / homology |  Function and homology information Function and homology informationviral release from host cell by cytolysis / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / host cell cytoplasm / defense response to bacterium Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Liu, L. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2009 Title: Use of stabilizing mutations to engineer a charged group within a ligand-binding hydrophobic cavity in T4 lysozyme. Authors: Liu, L. / Baase, W.A. / Michael, M.M. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3guj.cif.gz | 53.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3guj.ent.gz | 37.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3guj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gu/3gujftp://data.pdbj.org/pub/pdb/validation_reports/gu/3guj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3guiSC  3gukC  3gulC  3gumC  3gunC  3guoC  3gupC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 18709.568 Da / Num. of mol.: 1 / Mutation: T21C/S38D/L99A/M102E/E108V/S117V/T142C/N144D Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage T4 (virus) / Gene: E / Plasmid: p1403 / Production host:  |
|---|
-Non-polymers , 6 types, 183 molecules 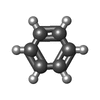





| #2: Chemical | ChemComp-BNZ /  Mass: 78.112 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6 Mass: 78.112 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6 |
|---|---|
| #3: Chemical | ChemComp-BME /  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS |
| #4: Chemical | ChemComp-CO3 /  Mass: 60.009 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO3 Mass: 60.009 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO3 |
| #5: Chemical | ChemComp-EPE /  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM |
| #6: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #7: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.04 Å3/Da / Density % sol: 39.81 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 30% PEG 8000, 0.2M ammounium sulfate, 0.1M Tris, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.2 / Wavelength: 1 Å / Beamline: 8.2.2 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Oct 7, 2007 / Details: Si(111) |
| Radiation | Monochromator: KOHZU / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→45.644 Å / Num. obs: 19653 / % possible obs: 92.5 % / Observed criterion σ(I): 2 / Redundancy: 6 % / Rmerge(I) obs: 0.065 / Net I/σ(I): 19.1 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 6.2 % / Rmerge(I) obs: 0.398 / Mean I/σ(I) obs: 3.9 / % possible all: 96.2 |
- Processing
Processing
| Software | Name: REFMAC / Version: 5.2.0019 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3GUI Resolution: 1.6→45.64 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.945 / SU B: 5.023 / SU ML: 0.08 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.118 / ESU R Free: 0.114 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.532 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→45.64 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.645 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
