 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3g46: Ligand migration and cavities within scapharca dimeric hemoglobin... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3g46 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ligand migration and cavities within scapharca dimeric hemoglobin: wild type with co bound to heme and chloroform bound to the XE4 cavity | ||||||
 Components Components | Globin-1 | ||||||
 Keywords Keywords | OXYGEN BINDING / OXYGEN TRANSPORT / ALLOSTERY / OXYGEN AFFINITY / CYTOPLASM / HEME / IRON / METAL-BINDING / OXYGEN STORAGE/TRANSPORT / Transport | ||||||
| Function / homology |  Function and homology information Function and homology informationoxygen carrier activity / oxygen binding / heme binding / metal ion binding / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Scapharca inaequivalvis (ark clam) Scapharca inaequivalvis (ark clam) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.91 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.91 Å | ||||||
 Authors Authors | Knapp, J.E. / Pahl, R. / Cohen, J. / Nichols, J.C. / Schulten, K. / Gibson, Q.H. / Srajer, V. / Royer Jr., W.E. | ||||||
 Citation Citation | Journal: Structure / Year: 2009 Title: Ligand migration and cavities within Scapharca Dimeric HbI: studies by time-resolved crystallo-graphy, Xe binding, and computational analysis. Authors: Knapp, J.E. / Pahl, R. / Cohen, J. / Nichols, J.C. / Schulten, K. / Gibson, Q.H. / Srajer, V. / Royer, W.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3g46.cif.gz | 155 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3g46.ent.gz | 121.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3g46.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g4/3g46ftp://data.pdbj.org/pub/pdb/validation_reports/g4/3g46 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3g4qC  3g4rC  3g4uC  3g4vC  3g4wC  3g4yC  3g52C  3g53C  3sdhS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | BIOMOLECULE: 1 SEE REMARK 350 FOR THE AUTHOR PROVIDED AND PROGRAM GENERATED ASSEMBLY INFORMATION FOR THE STRUCTURE IN THIS ENTRY. THE REMARK MAY ALSO PROVIDE INFORMATION ON BURIED SURFACE AREA. COORDINATES FOR A COMPLETE MULTIMER REPRESENTING THE KNOWN BIOLOGICALLY SIGNIFICANT OLIGOMERIZATION STATE OF THE MOLECULE CAN BE GENERATED BY APPLYING BIOMT TRANSFORMATIONS GIVEN BELOW. BOTH NON-CRYSTALLOGRAPHIC AND CRYSTALLOGRAPHIC OPERATIONS ARE GIVEN. APPLY THE FOLLOWING TO CHAINS: A, B BIOMT1 1 1.000000 0.000000 0.000000 0.00000 BIOMT2 1 0.000000 1.000000 0.000000 0.00000 BIOMT3 1 0.000000 0.000000 1.000000 0.00000 |
-Components
| #1: Protein | Mass: 15967.304 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Scapharca inaequivalvis (ark clam) / Gene: HBI / Plasmid: PCS-26 / Production host:  #2: Chemical |   Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4#3: Chemical | 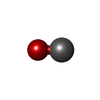  Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO#4: Chemical | ChemComp-XE /   Mass: 131.293 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Xe Mass: 131.293 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Xe#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 432 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 432 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.12 Å3/Da / Density % sol: 42 % |
|---|---|
| Crystal grow | Temperature: 296 K / Method: microbatch / pH: 7.5 Details: 1.5-2.5M PHOSPHATE BUFFER, PH 7.50, SMALL TUBES, TEMPERATURE 298K, Microbatch, temperature 296K |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 0.9 / Wavelength: 0.9 Å / Beamline: 14-BM-C / Wavelength: 0.9 / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 7, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 0.91→46.3 Å / Num. all: 205646 / Num. obs: 160199 / % possible obs: 77.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4 % / Rmerge(I) obs: 0.075 / Rsym value: 0.075 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 0.91→1.01 Å / Redundancy: 2.1 % / Rmerge(I) obs: 0.189 / Mean I/σ(I) obs: 4.7 / Rsym value: 0.189 / % possible all: 59.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3SDH Resolution: 0.91→46.3 Å / Num. parameters: 26633 / Num. restraintsaints: 33906 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY ?
| |||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.155 Å / Num. disordered residues: 34 / Occupancy sum hydrogen: 2315.44 / Occupancy sum non hydrogen: 2761.87 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.91→46.3 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
