Entry Database : PDB / ID : 3fy0Title Crystal structure of PAK1 kinase domain with ruthenium complex DW1 Serine/threonine-protein kinase PAK 1 Keywords / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 2.35 Å Authors Maksimoska, J. / Marmorstein, R. / Meggers, E. Journal : J.Am.Chem.Soc. / Year : 2008Title : Targeting Large Kinase Active Site with Rigid, Bulky Octahedral Ruthenium ComplexesAuthors : Maksimoska, J. / Feng, L. / Harms, K. / Yi, C. / Kissil, J. / Marmorstein, R. / Meggers, E. History Deposition Jan 21, 2009 Deposition site / Processing site Revision 1.0 Mar 3, 2009 Provider / Type Revision 1.1 Jul 13, 2011 Group / Version format complianceRevision 1.2 Nov 1, 2017 Group / Category / Item Revision 1.3 Oct 20, 2021 Group / Derived calculationsCategory database_2 / struct_conn ... database_2 / struct_conn / struct_ref_seq_dif / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_leaving_atom_flag / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.4 Nov 6, 2024 Group / Structure summaryCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / pdbx_entry_details / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
















 Assembly
Assembly

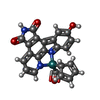
 Mass: 496.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H13N3O4Ru
Mass: 496.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H13N3O4Ru Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 119 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 23-ID-B
/ Beamline: 23-ID-B Processing
Processing