 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fx8: Distinct recognition of three-way DNA junctions by a thioester va... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fx8 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Distinct recognition of three-way DNA junctions by a thioester variant of a metallo-supramolecular cylinder ('helicate') | ||||||||||||||||||
 Components Components | (5'-D(* Keywords KeywordsDNA / Self-assembly / DNA-based nanomaterial | Function / homology | Chem-5PM / : / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.44 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.44 Å  Authors AuthorsBoer, D.R. / Uson, I. / Coll, M. |  CitationJournal: Angew.Chem.Int.Ed.Engl. / Year: 2010 CitationJournal: Angew.Chem.Int.Ed.Engl. / Year: 2010Title: Self-Assembly of Functionalizable Two-Component 3D DNA Arrays through the Induced Formation of DNA Three-Way-Junction Branch Points by Supramolecular Cylinders. Authors: Boer, D.R. / Kerckhoffs, J.M. / Parajo, Y. / Pascu, M. / Uson, I. / Lincoln, P. / Hannon, M.J. / Coll, M. #1: Journal: Angew.Chem.Int.Ed.Engl. / Year: 2006Title: Molecular recognition of a three-way DNA junction by a metallosupramolecular helicate. Authors: Oleksy, A. / Blanco, A.G. / Boer, R. / Uson, I. / Aymami, J. / Rodger, A. / Hannon, M.J. / Coll, M. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fx8.cif.gz | 15.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fx8.ent.gz | 9.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3fx8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fx/3fx8ftp://data.pdbj.org/pub/pdb/validation_reports/fx/3fx8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3i1dC  3fx9 C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 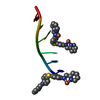
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||
| Details | A THREE-WAY JUNCTION (CHAINS A AND C) WITH DISCONTINUOUS DNA STRANDS CAN BE GENERATED THROUGH THE FOLLOWING TRANSFORMATIONS ON CHAIN C: [Z+1/2,-X+1/2,-Y+(0 1 1)], [Y+1/2,-Z+1/2,-X+(-1 1 1)], [-X, Y+1/2,-Z+1/2+(0 1 0)], [-Z,X+1/2,-Y+1/2+(0 1 1)], [Y,Z,X+(0 2 1)] AND THE FOLLOWING TRANSFORMATIONS ON CHAIN A: [-Z,X+1/2,-Y+1/2 +(0 1 1)], [Y+1/2,-Z+1/2,-X +(-1 1 1)] |
-Components
| #1: DNA chain | ( Mass: 1809.217 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: Synthetic hexanucleotide. The sequence is palindromic | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-FE2 /   Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe#3: Chemical | 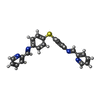  Mass: 394.492 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H18N4S Mass: 394.492 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H18N4SNonpolymer details | LIGAND 5PM C 111 WITH TWO IRON ATOMS FE2 C 101 AND C 102 IS THE M HELICAL ENANTIOMER OF [FE2L3], ...LIGAND 5PM C 111 WITH TWO IRON ATOMS FE2 C 101 AND C 102 IS THE M HELICAL ENANTIOMER | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.5 Å3/Da / Density % sol: 77.64 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 1 microliter of 10 mM cylinder, 1 microliter of 3 mM DNA, 2 microliters of crystallization buffer (10 mM Magnesium chloride, 5% v/v Isopropanol, 50 mM Tris-HCl), pH 8.5, VAPOR DIFFUSION, ...Details: 1 microliter of 10 mM cylinder, 1 microliter of 3 mM DNA, 2 microliters of crystallization buffer (10 mM Magnesium chloride, 5% v/v Isopropanol, 50 mM Tris-HCl), pH 8.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K | ||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 1.7366, 1.7410, 1.7311 / Beamline: ID29 / Wavelength: 1.7366, 1.7410, 1.7311 | ||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Feb 23, 2006 | ||||||||||||
| Radiation | Monochromator: Si(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.44→24.63 Å / Num. all: 1573 / Num. obs: 1573 / % possible obs: 99.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 8.4 % / Biso Wilson estimate: 94 Å2 / Rmerge(I) obs: 0.041 / Net I/σ(I): 36.1 | ||||||||||||
| Reflection shell | Resolution: 2.44→2.58 Å / Redundancy: 5.1 % / Rmerge(I) obs: 0.447 / Mean I/σ(I) obs: 2.2 / Num. unique all: 228 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.44→24.63 Å / Isotropic thermal model: isotropic
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET (SHELXL SWAT) | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 79.4 Å2 | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.44→24.63 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.44→2.55 Å /
|
