 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3bif: 6-PHOSPHOFRUCTO-2-KINASE/FRUCTOSE-2,6-BISPHOSPHATASE EMPTY 6-PF-2... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bif | ||||||
|---|---|---|---|---|---|---|---|
| Title | 6-PHOSPHOFRUCTO-2-KINASE/FRUCTOSE-2,6-BISPHOSPHATASE EMPTY 6-PF-2K ACTIVE SITE | ||||||
 Components Components | PROTEIN (6-PHOSPHOFRUCTO-2-KINASE/ FRUCTOSE-2,6-BISPHOSPHATASE) | ||||||
 Keywords Keywords | TRANSFERASE / KINASE / PHOSPHOTRANSFERASE / PHOSPHATASE / HYDROLASE (PHOSPHO) / GLYCOLYSIS / BIFUNCTIONAL ENZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationRegulation of glycolysis by fructose 2,6-bisphosphate metabolism / 6-phosphofructo-2-kinase / 6-phosphofructo-2-kinase activity / fructose 2,6-bisphosphate metabolic process / fructose-2,6-bisphosphate 2-phosphatase / fructose-2,6-bisphosphate 2-phosphatase activity / fructose metabolic process / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Yuen, M.H. / Hasemann, C.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: A switch in the kinase domain of rat testis 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Authors: Yuen, M.H. / Wang, X.L. / Mizuguchi, H. / Uyeda, K. / Hasemann, C.A. #1: Journal: Structure / Year: 1996Title: The crystal structure of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase reveals distinct domain homologies. Authors: Hasemann, C.A. / Istvan, E.S. / Uyeda, K. / Deisenhofer, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bif.cif.gz | 108.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bif.ent.gz | 82 KB | Display | PDB format |
| PDBx/mmJSON format | 3bif.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bi/3bifftp://data.pdbj.org/pub/pdb/validation_reports/bi/3bif | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bifS S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53937.387 Da / Num. of mol.: 1 / Mutation: W15F, W64F, W299F, W320F Source method: isolated from a genetically manipulated source Details: BIFUNCTIONAL ENZYME, ALSO EC 3.1.3.46 / Source: (gene. exp.)  Keywords: BIFUNCTIONAL ENZYME, ALSO EC 3.1.3.46 / References: UniProt: P25114, 6-phosphofructo-2-kinase Keywords: BIFUNCTIONAL ENZYME, ALSO EC 3.1.3.46 / References: UniProt: P25114, 6-phosphofructo-2-kinase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Sugar | 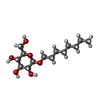  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H28O6 / Comment: detergent*YM Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H28O6 / Comment: detergent*YM#3: Chemical |   Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#4: Chemical | ChemComp-SIN / | 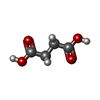  Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 187 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 187 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 57.6 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: PROTEIN WAS CRYSTALLIZED FROM 50MM SUCCINATE, PH 5.5, 50MM TRIS-PO4, PH 7.5, 11% PEG 4000, 100UM EDTA, 10MM DTT, 10% GLYCEROL, 3MM MGCL2., pH 7.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Oct 9, 1997 / Details: MIRRORS |
| Radiation | Monochromator: MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→30 Å / Num. obs: 23883 / % possible obs: 88.1 % / Observed criterion σ(I): 1 / Redundancy: 2.64 % / Biso Wilson estimate: 39.9 Å2 / Rsym value: 0.039 / Net I/σ(I): 36.5 |
| Reflection shell | Resolution: 2.3→2.35 Å / Redundancy: 2.3 % / Mean I/σ(I) obs: 7.16 / Rsym value: 0.15 / % possible all: 84.9 |
| Reflection | *PLUS Rmerge(I) obs: 0.039 |
| Reflection shell | *PLUS % possible obs: 84.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1BIF Resolution: 2.3→30 Å / Rfactor Rfree error: 0.0049 / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.0001 / Isotropic thermal model: RESTRAINED / σ(F): 1 Details: THE N-TERMINAL 36 AMINO ACIDS OF THE PROTEIN WERE NOT VISIBLE IN THE ELECTRON DENSITY. PROTEIN SEQUENCE ANALYSIS OF A REDISSOLVED CRYSTAL CONFIRMED THE PRESENCE OF THE N-TERMINUS IN THE ...Details: THE N-TERMINAL 36 AMINO ACIDS OF THE PROTEIN WERE NOT VISIBLE IN THE ELECTRON DENSITY. PROTEIN SEQUENCE ANALYSIS OF A REDISSOLVED CRYSTAL CONFIRMED THE PRESENCE OF THE N-TERMINUS IN THE CRYSTALS. THE CONCLUSION IS THAT THE N-TERMINAL 36 AMINO ACIDS ARE DISORDERED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.4 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: ENGH & HUBER / Topol file: ENGH & HUBER | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.3 Å / Lowest resolution: 30 Å / σ(F): 1 / Rfactor obs: 0.197 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 39 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.358 / % reflection Rfree: 10 % / Rfactor Rwork: 0.319 |
