- PDB-2yzj: Crystal structure of dCTP deaminase from Sulfolobus tokodaii -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 2yzj
タイトル
Crystal structure of dCTP deaminase from Sulfolobus tokodaii
要素
167aa long hypothetical dUTPase
キーワード
STRUCTURAL GENOMICS / UNKNOWN FUNCTION / All beta proteins / Hypothetical protein / NPPSFA / National Project on Protein Structural and Functional Analyses / RIKEN Structural Genomics/Proteomics Initiative / RSGI
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Sulfolobus tokodaii (古細菌)
Sulfolobus tokodaii (古細菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体

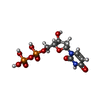
 分子量: 388.162 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C9H14N2O11P2
分子量: 388.162 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C9H14N2O11P2
 分子量: 122.143 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM
分子量: 122.143 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM
 分子量: 24.305 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Mg 分子量: 18.015 Da / 分子数: 299 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 299 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: BL26B2 / 波長: 0.9789, 0.9794, 0.9000
/ ビームライン: BL26B2 / 波長: 0.9789, 0.9794, 0.9000 解析
解析