| Entry | Database: PDB / ID: 2ykx
|
|---|
| Title | Structural Determinants of the Beta-Selectivity of a Bacterial Aminotransferase |
|---|
 Components Components | BETA-TRANSAMINASE |
|---|
 Keywords Keywords | TRANSFERASE |
|---|
| Function / homology |  Function and homology information Function and homology information
Aminotransferase class-III / Aminotransferase class-III / Aspartate Aminotransferase, domain 1 / Aspartate Aminotransferase, domain 1 / Aspartate Aminotransferase; domain 2 / Type I PLP-dependent aspartate aminotransferase-like (Major domain) / Pyridoxal phosphate-dependent transferase, small domain / Pyridoxal phosphate-dependent transferase, major domain / Pyridoxal phosphate-dependent transferase / Alpha-Beta Complex ...Aminotransferase class-III / Aminotransferase class-III / Aspartate Aminotransferase, domain 1 / Aspartate Aminotransferase, domain 1 / Aspartate Aminotransferase; domain 2 / Type I PLP-dependent aspartate aminotransferase-like (Major domain) / Pyridoxal phosphate-dependent transferase, small domain / Pyridoxal phosphate-dependent transferase, major domain / Pyridoxal phosphate-dependent transferase / Alpha-Beta Complex / 3-Layer(aba) Sandwich / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  MESORHIZOBIUM SP. LUK (bacteria) MESORHIZOBIUM SP. LUK (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å |
|---|
 Authors Authors | Wybenga, G.G. / Crismaru, C.G. / Janssen, D.B. / Dijkstra, B.W. |
|---|
 Citation Citation | Journal: J.Biol.Chem. / Year: 2012
Title: Structural Determinants of the Beta-Selectivity of a Bacterial Aminotransferase.
Authors: Wybenga, G.G. / Crismaru, C.G. / Janssen, D.B. / Dijkstra, B.W. |
|---|
| History | | Deposition | May 30, 2011 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | May 30, 2012 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 11, 2012 | Group: Other |
|---|
| Revision 1.2 | Aug 29, 2012 | Group: Database references |
|---|
| Revision 1.3 | May 1, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.pdbx_leaving_atom_flag / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj

 Assembly
Assembly



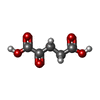



 Mass: 247.142 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H10NO6P
Mass: 247.142 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 146.098 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C5H6O5
Mass: 146.098 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing