GET complex / : / tail-anchored membrane protein insertion into ER membrane / Hydrolases; Acting on acid anhydrides / protein insertion into ER membrane / post-translational protein targeting to endoplasmic reticulum membrane / response to arsenic-containing substance / response to metal ion / retrograde vesicle-mediated transport, Golgi to endoplasmic reticulum / protein folding chaperone ...GET complex / : / tail-anchored membrane protein insertion into ER membrane / Hydrolases; Acting on acid anhydrides / protein insertion into ER membrane / post-translational protein targeting to endoplasmic reticulum membrane / response to arsenic-containing substance / response to metal ion / retrograde vesicle-mediated transport, Golgi to endoplasmic reticulum / protein folding chaperone / guanyl-nucleotide exchange factor activity / : / response to heat / cellular response to oxidative stress / endoplasmic reticulum membrane / Golgi apparatus / endoplasmic reticulum / ATP hydrolysis activity / ATP binding / metal ion binding / identical protein binding / cytosol Similarity search - Function
Type: MARRESEARCH / Detector: CCD / Date: Aug 10, 2008
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.97856 Å / Relative weight: 1
Reflection
Resolution: 2→50 Å / Num. obs: 94446 / % possible obs: 95.9 % / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 21.1
Reflection shell
Resolution: 2→2.07 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.59 / Mean I/σ(I) obs: 1.9 / % possible all: 86.3
-
Processing
Software
Name
Version
Classification
PHENIX
(PHENIX.REFINE)
refinement
DENZO
datareduction
SCALEPACK
datascaling
PHASER
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: PARTIAL MODEL DERIVED FROM SAD DATASET Resolution: 1.994→37.441 Å / SU ML: 0 / σ(F): 1.34 / Phase error: 21.7 / Stereochemistry target values: ML Details: DISORDERED SIDECHAINS WERE MODELED STEREOCHEMICALLY.
Rfactor
Num. reflection
% reflection
Rfree
0.2127
4715
5 %
Rwork
0.1759
-
-
obs
0.1778
94446
95.75 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 57.944 Å2 / ksol: 0.363 e/Å3
Displacement parameters
Biso mean: 47.39 Å2
Baniso -1
Baniso -2
Baniso -3
1-
3.9173 Å2
0 Å2
-8.7466 Å2
2-
-
0.0133 Å2
-0 Å2
3-
-
-
-3.9307 Å2
Refinement step
Cycle: LAST / Resolution: 1.994→37.441 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
9368
0
134
512
10014
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.011
9694
X-RAY DIFFRACTION
f_angle_d
1.331
13068
X-RAY DIFFRACTION
f_dihedral_angle_d
17.385
3639
X-RAY DIFFRACTION
f_chiral_restr
0.088
1485
X-RAY DIFFRACTION
f_plane_restr
0.006
1641
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
1.9937-2.0164
0.2879
115
0.2542
2452
X-RAY DIFFRACTION
78
2.0164-2.0401
0.281
154
0.2366
2690
X-RAY DIFFRACTION
88
2.0401-2.065
0.2739
150
0.2288
2729
X-RAY DIFFRACTION
87
2.065-2.0911
0.2475
125
0.2157
2892
X-RAY DIFFRACTION
93
2.0911-2.1186
0.2545
150
0.2088
2856
X-RAY DIFFRACTION
92
2.1186-2.1477
0.2593
163
0.1981
2863
X-RAY DIFFRACTION
92
2.1477-2.1783
0.2292
165
0.195
2907
X-RAY DIFFRACTION
94
2.1783-2.2108
0.2361
175
0.1863
2832
X-RAY DIFFRACTION
94
2.2108-2.2454
0.2237
134
0.1757
2958
X-RAY DIFFRACTION
93
2.2454-2.2822
0.2337
160
0.1784
2910
X-RAY DIFFRACTION
94
2.2822-2.3215
0.2147
160
0.1764
2918
X-RAY DIFFRACTION
94
2.3215-2.3638
0.2227
147
0.176
2928
X-RAY DIFFRACTION
94
2.3638-2.4092
0.2209
165
0.1718
2984
X-RAY DIFFRACTION
96
2.4092-2.4584
0.2149
167
0.1754
3000
X-RAY DIFFRACTION
96
2.4584-2.5118
0.2498
163
0.1771
3003
X-RAY DIFFRACTION
97
2.5118-2.5702
0.2141
150
0.1696
3052
X-RAY DIFFRACTION
97
2.5702-2.6345
0.201
137
0.1641
3078
X-RAY DIFFRACTION
99
2.6345-2.7057
0.2158
165
0.164
3127
X-RAY DIFFRACTION
99
2.7057-2.7853
0.213
148
0.1669
3104
X-RAY DIFFRACTION
99
2.7853-2.8752
0.2113
156
0.1721
3106
X-RAY DIFFRACTION
99
2.8752-2.9779
0.2423
163
0.1794
3125
X-RAY DIFFRACTION
99
2.9779-3.0971
0.2044
164
0.1689
3099
X-RAY DIFFRACTION
100
3.0971-3.2379
0.2167
176
0.1619
3082
X-RAY DIFFRACTION
100
3.2379-3.4085
0.18
159
0.1584
3154
X-RAY DIFFRACTION
100
3.4085-3.6219
0.1966
161
0.1515
3112
X-RAY DIFFRACTION
100
3.6219-3.9013
0.1859
165
0.1523
3136
X-RAY DIFFRACTION
100
3.9013-4.2934
0.1833
163
0.1592
3129
X-RAY DIFFRACTION
100
4.2934-4.9135
0.199
171
0.1567
3165
X-RAY DIFFRACTION
100
4.9135-6.186
0.2182
170
0.1873
3148
X-RAY DIFFRACTION
100
6.186-37.4475
0.2087
174
0.2058
3192
X-RAY DIFFRACTION
99
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
1.6791
-0.5233
0.4978
2.2263
0.0811
2.5767
0.1969
0.2253
-0.1987
-0.3887
-0.1808
0.0912
0.1213
-0.0024
-0.0001
0.2198
0.1164
0.0291
0.2267
-0.0377
0.1664
15.0242
38.9704
49.9513
2
1.6772
-0.5493
0.3718
1.7612
-0.2109
2.6288
0.0091
-0.1779
-0.0166
0.1802
-0.0893
-0.0865
-0.2196
0.0692
-0.0001
0.1495
-0.0183
0.0654
0.1915
0.0048
0.1508
22.1913
46.2015
73.5889
3
2.3496
-0.4854
0.5835
2.2697
0.2174
1.9753
-0.0383
0.1494
-0.1345
-0.0331
0.1088
0.068
0.2077
0.0218
0
0.2381
0.0576
-0.0202
0.2033
-0.0346
0.2004
47.7993
-13.2446
18.7067
4
1.9476
-0.1593
0.2964
1.637
0.2593
1.9044
-0.2533
-0.0149
0.411
0.0753
0.1377
0.0827
-0.3389
-0.1133
-0
0.3423
0.0916
-0.0971
0.2291
-0.0346
0.3742
41.2242
10.5908
23.3607
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
CHAINA
2
X-RAY DIFFRACTION
2
CHAINB
3
X-RAY DIFFRACTION
3
CHAINC
4
X-RAY DIFFRACTION
4
CHAIND
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj




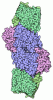

 Assembly
Assembly




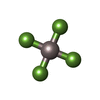



 Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 102.975 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: AlF4
Mass: 102.975 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: AlF4 Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Sample preparation
Sample preparation / Beamline: 21-ID-G / Wavelength: 0.97856
/ Beamline: 21-ID-G / Wavelength: 0.97856  Processing
Processing