 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vdc: THE 9.5 A RESOLUTION STRUCTURE OF GLUTAMATE SYNTHASE FROM CRYO-EL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vdc | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE 9.5 A RESOLUTION STRUCTURE OF GLUTAMATE SYNTHASE FROM CRYO-ELECTRON MICROSCOPY AND ITS OLIGOMERIZATION BEHAVIOR IN SOLUTION: FUNCTIONAL IMPLICATIONS. | ||||||
 Components Components | (GLUTAMATE SYNTHASE [NADPH] ...) x 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / AMIDOTRANSFERASE / AMMONIA ASSIMILATION / NADP / IRON / ZYMOGEN / NADPH-DEPENDENT GLUTAMATE SYNTHASE / IRON SULPHUR FLAVOPROTEIN / GLUTAMINE AMIDOTRANSFERASE / GLUTAMATE BIOSYNTHESIS / AMINO-ACID BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationglutamate synthase (NADPH) / glutamate synthase (NADPH) activity / L-glutamate biosynthetic process / ammonia assimilation cycle / 3 iron, 4 sulfur cluster binding / 4 iron, 4 sulfur cluster binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  AZOSPIRILLUM BRASILENSE (bacteria) AZOSPIRILLUM BRASILENSE (bacteria) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 9.5 Å | ||||||
 Authors Authors | Cottevieille, M. / Larquet, E. / Jonic, S. / Petoukhov, M.V. / Caprini, G. / Paravisi, S. / Svergun, D.I. / Vanoni, M.A. / Boisset, N. | ||||||
 Citation Citation | Journal: J Biol Chem / Year: 2008 Title: The subnanometer resolution structure of the glutamate synthase 1.2-MDa hexamer by cryoelectron microscopy and its oligomerization behavior in solution: functional implications. Authors: Magali Cottevieille / Eric Larquet / Slavica Jonic / Maxim V Petoukhov / Gianluca Caprini / Stefano Paravisi / Dmitri I Svergun / Maria A Vanoni / Nicolas Boisset /  Abstract: The three-dimensional structure of the hexameric (alphabeta)(6) 1.2-MDa complex formed by glutamate synthase has been determined at subnanometric resolution by combining cryoelectron microscopy, ...The three-dimensional structure of the hexameric (alphabeta)(6) 1.2-MDa complex formed by glutamate synthase has been determined at subnanometric resolution by combining cryoelectron microscopy, small angle x-ray scattering, and molecular modeling, providing for the first time a molecular model of this complex iron-sulfur flavoprotein. In the hexameric species, interprotomeric alpha-alpha and alpha-beta contacts are mediated by the C-terminal domain of the alpha subunit, which is based on a beta helical fold so far unique to glutamate synthases. The alphabeta protomer extracted from the hexameric model is fully consistent with it being the minimal catalytically active form of the enzyme. The structure clarifies the electron transfer pathway from the FAD cofactor on the beta subunit, to the FMN on the alpha subunit, through the low potential [4Fe-4S](1+/2+) centers on the beta subunit and the [3Fe-4S](0/1+) cluster on the alpha subunit. The (alphabeta)(6) hexamer exhibits a concentration-dependent equilibrium with alphabeta monomers and (alphabeta)(2) dimers, in solution, the hexamer being destabilized by high ionic strength and, to a lower extent, by the reaction product NADP(+). Hexamerization seems to decrease the catalytic efficiency of the alphabeta protomer only 3-fold by increasing the K(m) values measured for l-Gln and 2-OG. However, it cannot be ruled out that the (alphabeta)(6) hexamer acts as a scaffold for the assembly of multienzymatic complexes of nitrogen metabolism or that it provides a means to regulate the activity of the enzyme through an as yet unknown ligand. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AJ" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AJ" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BH" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "CJ" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "DH" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "EJ" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "FH" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vdc.cif.gz | 2 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vdc.ent.gz | 1.6 MB | Display | PDB format |
| PDBx/mmJSON format | 2vdc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vd/2vdcftp://data.pdbj.org/pub/pdb/validation_reports/vd/2vdc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1440MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
-GLUTAMATE SYNTHASE [NADPH] ... , 2 types, 12 molecules ABCDEFGHIJKL
| #1: Protein | Mass: 161615.875 Da / Num. of mol.: 6 / Fragment: RESIDUES 37-1508, ALPHA SUBUNIT Source method: isolated from a genetically manipulated source Source: (gene. exp.) AZOSPIRILLUM BRASILENSE (bacteria) / Strain: SP7 / Plasmid: PET11A / Production host: #2: Protein | Mass: 49367.531 Da / Num. of mol.: 6 / Fragment: RESIDUES 27-482, BETA SUBUNIT Source method: isolated from a genetically manipulated source Source: (gene. exp.) AZOSPIRILLUM BRASILENSE (bacteria) / Strain: SP7 / Plasmid: PET11A / Production host: |
|---|
-Non-polymers , 6 types, 42 molecules 
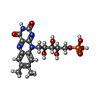
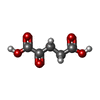
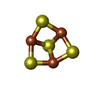


| #3: Chemical | ChemComp-OMT /  Type: L-peptide linking / Mass: 181.210 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C5H11NO4S Type: L-peptide linking / Mass: 181.210 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C5H11NO4S#4: Chemical | ChemComp-FMN /  Mass: 456.344 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C17H21N4O9P#5: Chemical | ChemComp-AKG /  Mass: 146.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C5H6O5#6: Chemical | ChemComp-F3S /  Mass: 295.795 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe3S4 Mass: 295.795 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe3S4#7: Chemical | ChemComp-SF4 /  Mass: 351.640 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 351.640 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Fe4S4#8: Chemical | ChemComp-FAD /  Mass: 785.550 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: AZOSPIRILLUM BRASILENSE GLUTAMATE SYNTHASE / Type: COMPLEX |
|---|---|
| Buffer solution | Name: 25 MM HEPES/KOH, 1 MM EDTA, 1 MM DTT / pH: 7.5 / Details: 25 MM HEPES/KOH, 1 MM EDTA, 1 MM DTT |
| Specimen | Conc.: 9.25 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Details: HOLEY CARBON |
| Vitrification | Instrument: HOMEMADE PLUNGER / Cryogen name: ETHANE / Details: NLIQUID ETHANE |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: JEOL 2010UHR / Date: May 10, 2005 Details: CC =1.1 MM, FOCAL -LEN-1.9 M. MICROGRAPH WITH ANISOTROPIC OR NO DIFFRACTION RINGS WERE REMOVED USING THE ENHANCED POWER SPECTRA SORTING METHOD (JONIC S,SORZANO CO,COTTEVIEILLE M, LARQUET E, ...Details: CC =1.1 MM, FOCAL -LEN-1.9 M. MICROGRAPH WITH ANISOTROPIC OR NO DIFFRACTION RINGS WERE REMOVED USING THE ENHANCED POWER SPECTRA SORTING METHOD (JONIC S,SORZANO CO,COTTEVIEILLE M, LARQUET E, BOISSET N.,A NOVEL METHOD FOR IMPROVEMENT OF VISUALIZATION OF POWER SPECTRA FOR SORTING CRYO-ELECTRON MICROGRAPHS AND THEIR LOCAL AREAS. J STRUCT BIOL. 2007, 157(1),156-67.) |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 50000 X / Nominal defocus max: 3200 nm / Nominal defocus min: 1700 nm / Cs: 0.5 mm |
| Specimen holder | Temperature: 93.15 K / Tilt angle max: 0 ° / Tilt angle min: 0 ° |
| Image recording | Electron dose: 10 e/Å2 / Film or detector model: KODAK SO-163 FILM |
| Image scans | Num. digital images: 151 |
| Radiation wavelength | Relative weight: 1 |
- Processing
Processing
| EM software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Details: WIENER FILTERING OF VOLUMES FROM FOCAL SERIES | ||||||||||||
| Symmetry | Point symmetry: I (icosahedral) | ||||||||||||
| 3D reconstruction | Method: RANDOM CONICAL TILT SERIES (CRYOEM) AND REFINEMENT BY PROJECTION MATCHING AND CTF CORRECTION USING WIENER FILTERING OF VOLUMES FROM FOCAL SERIES Resolution: 9.5 Å / Num. of particles: 12800 / Nominal pixel size: 1.59 Å Details: THE CHAINS A,B,C,D,E,F OF THE COMPLEX CORRESPOND TO THE ALPHA SUBUNITS, SOLVED BY X-RAY (PDB CODE 1EA0). THREE DISORDERED REGIONS OF THIS X-RAY STRUCTURE (CHAINS A,B,C,D,E, F), IE RESIDUES ...Details: THE CHAINS A,B,C,D,E,F OF THE COMPLEX CORRESPOND TO THE ALPHA SUBUNITS, SOLVED BY X-RAY (PDB CODE 1EA0). THREE DISORDERED REGIONS OF THIS X-RAY STRUCTURE (CHAINS A,B,C,D,E, F), IE RESIDUES 305-307, 1172-1179 AND 1194-1202 WERE MODELLED WITH MODELLER. THE CHAINS G,H,I,J,K,L CORRESPOND TO THE HOMOLOGY MODEL OF THE BETA SUBUNIT (GENERATED BY MODELLER). Symmetry type: POINT | ||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT / Space: REAL Details: METHOD--REAL-SPACE LOCAL OPTIMIZATION REFINEMENT PROTOCOL--X-RAY AND HOMOLOGY MODELLING | ||||||||||||
| Atomic model building | PDB-ID: 2VDC Accession code: 2VDC / Source name: PDB / Type: experimental model | ||||||||||||
| Refinement | Highest resolution: 9.5 Å | ||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 9.5 Å
|
