- PDB-2vbk: NATIVE TAILSPIKE PROTEIN OF BACTERIOPHAGE SF6 -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2vbk
Title
NATIVE TAILSPIKE PROTEIN OF BACTERIOPHAGE SF6
Components
TAILSPIKE-PROTEIN
Keywords
VIRAL PROTEIN / VIRAL ADHESION PROTEIN / HYDROLASE / TAILSPIKE / ENDORHAMNOSIDASE / RIGHT-HANDED PARALLEL BETA-HELIX
Function / homology
Function and homology information
endo-1,3-alpha-L-rhamnosidase activity / symbiont entry into host cell via disruption of host cell envelope lipopolysaccharide / virus tail, fiber / symbiont entry into host cell via disruption of host cell envelope / symbiont entry into host / Hydrolases; Glycosylases; Glycosidases, i.e. enzymes that hydrolyse O- and S-glycosyl compounds / virion attachment to host cell Similarity search - Function
Resolution: 1.25→14.92 Å / Cor.coef. Fo:Fc: 0.979 / Cor.coef. Fo:Fc free: 0.974 / SU B: 0.478 / SU ML: 0.021 / Cross valid method: THROUGHOUT / ESU R: 0.031 / ESU R Free: 0.032 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. PROTEIN ATOM OCCUPANCIES LESS THAN 1.0 AND FOR WHICH NO ALTERNATE LOCATION ATOM RECORD IS PROVIDED ARE MOSTLY DUE TO RADIATION DAMAGES.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.142
8511
5 %
RANDOM
Rwork
0.119
-
-
-
obs
0.12
161221
97.4 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information ENTEROBACTERIA PHAGE SF6 (virus)
ENTEROBACTERIA PHAGE SF6 (virus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj
 Assembly
Assembly






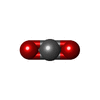

 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 62.068 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C2H6O2
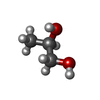
Mass: 62.068 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2
Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4 Mass: 44.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO2
Mass: 44.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO2 Sample preparation
Sample preparation / Beamline: BW7B / Wavelength: 0.842
/ Beamline: BW7B / Wavelength: 0.842  Processing
Processing