 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
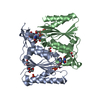
Yorodumi- PDB-2v9c: X-ray Crystallographic Structure of a Pseudomonas aeruginosa Azor... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2v9c | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray Crystallographic Structure of a Pseudomonas aeruginosa Azoreductase in Complex with Methyl Red. | ||||||
 Components Components | FMN-DEPENDENT NADH-AZOREDUCTASE 1 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FMN / NAD / FLAVODOXIN / FLAVOPROTEIN / NADPH-DEPENDENT / FLAVIN MONONUCLEOTIDE | ||||||
| Function / homology |  Function and homology information Function and homology informationquinone reductase (NADPH) activity / FMN-dependent NADH-azoreductase / oxidoreductase activity, acting on NAD(P)H as acceptor / Oxidoreductases; Acting on NADH or NADPH; With a quinone or similar compound as acceptor / oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor / FMN binding / response to oxidative stress / electron transfer activity Similarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.18 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.18 Å | ||||||
 Authors Authors | Wang, C.-J. / Hagemeier, C. / Rahman, N. / Lowe, E.D. / Noble, M.E.M. / Coughtrie, M. / Sim, E. / Westwood, I.M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2007 Title: Molecular Cloning, Characterisation and Ligand- Bound Structure of an Azoreductase from Pseudomonas Aeruginosa Authors: Wang, C.-J. / Hagemeier, C. / Rahman, N. / Lowe, E.D. / Noble, M.E.M. / Coughtrie, M. / Sim, E. / Westwood, I.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2v9c.cif.gz | 173.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2v9c.ent.gz | 138.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2v9c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v9/2v9cftp://data.pdbj.org/pub/pdb/validation_reports/v9/2v9c | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.2818, -0.7354, 0.6163), Vector: |
-Components
| #1: Protein | Mass: 23359.348 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: FMN AND METHYL RED COMPLEX / Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAO1 / Plasmid: PET28B / Production host: #2: Chemical | 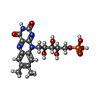  Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P#3: Chemical |   Mass: 269.299 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H15N3O2 Mass: 269.299 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H15N3O2#4: Chemical |   Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | 2-((4-(DIMETHYLAM | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.27 Å3/Da / Density % sol: 45.4 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 1.6 M AMMONIUM SULFATE, 0.1 M HEPES, PH 7.5. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9763 / Beamline: ID29 / Wavelength: 0.9763 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Apr 25, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 2.18→34 Å / Num. obs: 20681 / % possible obs: 97.4 % / Observed criterion σ(I): 1.6 / Redundancy: 3.5 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 9.7 |
| Reflection shell | Resolution: 2.18→2.3 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.46 / Mean I/σ(I) obs: 1.6 / % possible all: 88.8 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1V4B AND 1T5B Resolution: 2.18→34 Å / Stereochemistry target values: ML Details: RESIDUES -2 - 1 ARE DISORDERED. RESIDUES 125 - 126 AND 187 - 192 OF CHAIN A ARE DISORDERED. RESIDUES 123 - 128 AND 187 - 198 OF CHAIN B ARE DISORDERED. REFMAC VERSION 5.3.0037 WAS USED FOR ...Details: RESIDUES -2 - 1 ARE DISORDERED. RESIDUES 125 - 126 AND 187 - 192 OF CHAIN A ARE DISORDERED. RESIDUES 123 - 128 AND 187 - 198 OF CHAIN B ARE DISORDERED. REFMAC VERSION 5.3.0037 WAS USED FOR REFINEMENT EXCEPT FOR THE FINAL REFINEMENT STEP, FOR WHICH PHENIX.REFINE WAS USED.
| ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.18→34 Å
|
