 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2tmd: CORRELATION OF X-RAY DEDUCED AND EXPERIMENTAL AMINO ACID SEQUENCE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2tmd | ||||||
|---|---|---|---|---|---|---|---|
| Title | CORRELATION OF X-RAY DEDUCED AND EXPERIMENTAL AMINO ACID SEQUENCES OF TRIMETHYLAMINE DEHYDROGENASE | ||||||
 Components Components | TRIMETHYLAMINE DEHYDROGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtrimethylamine dehydrogenase / trimethylamine dehydrogenase activity / FMN binding / 4 iron, 4 sulfur cluster binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Methylophilus methylotrophus (bacteria) Methylophilus methylotrophus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.4 Å X-RAY DIFFRACTION / Resolution: 2.4 Å | ||||||
 Authors Authors | Mathews, F.S. / Lim, L.W. / White, S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1992 Title: Correlation of x-ray deduced and experimental amino acid sequences of trimethylamine dehydrogenase. Authors: Barber, M.J. / Neame, P.J. / Lim, L.W. / White, S. / Matthews, F.S. #1: Journal: J.Biol.Chem. / Year: 1989Title: Studies of Crystalline Trimethylamine Dehydrogenase in Three Oxidation States and in the Presence of Substrate and Inhibitor Authors: Bellamy, H.D. / Lim, L.W. / Mathews, F.S. / Dunham, W.R. #2: Journal: J.Biol.Chem. / Year: 1988Title: Identification of Adp in the Iron-Sulfur Flavoprotein Trimethylamine Dehydrogenase Authors: Lim, L.W. / Mathews, F.S. / Steenkamp, D.J. #3: Journal: J.Biol.Chem. / Year: 1986Title: Three-Dimensional Structure of the Iron-Sulfur Flavoprotein Trimethylamine Dehydrogenase at 2.4 Angstroms Resolution Authors: Lim, L.W. / Shamala, N. / Mathews, F.S. / Steenkamp, D.J. / Hamlin, R. / Xuong, N.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2tmd.cif.gz | 305.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2tmd.ent.gz | 245.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2tmd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tm/2tmdftp://data.pdbj.org/pub/pdb/validation_reports/tm/2tmd | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: THR A 70 - HIS A 71 OMEGA = 17.29 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: THR B 70 - HIS B 71 OMEGA = 16.18 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.971338, 0.217797, 0.095224), Vector: Details | THERE ARE TWO MOLECULES IN THE ASYMMETRIC UNIT. THE TRANSFORMATION PRESENTED ON *MTRIX* RECORDS BELOW WILL YIELD APPROXIMATE COORDINATES FOR CHAIN *B* WHEN APPLIED TO CHAIN *A*. | |
-Components
| #1: Protein | Mass: 81606.023 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methylophilus methylotrophus (bacteria)Strain: W3A1 / References: UniProt: P16099, EC: 1.5.99.7 #2: Chemical |   Mass: 351.640 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 351.640 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe4S4#3: Chemical | 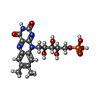  Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P#4: Chemical |   Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 581 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 581 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.51 % |
|---|---|
| Crystal grow | *PLUS pH: 6.5 / Method: vapor diffusion, hanging dropDetails: taken from Lim, L. et al (1982). J. Mol. Biol., 162, 869-876., macro-seeding |
| Components of the solutions | *PLUS Conc.: 10-12 % / Common name: PEG8000 |
-Data collection
| Reflection | *PLUS Highest resolution: 2.4 Å / Num. obs: 133000 / Num. measured all: 430000 / Rmerge(I) obs: 0.032 |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.4→10 Å / Rfactor Rwork: 0.154 / Rfactor obs: 0.154 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 10 Å / Rfactor obs: 0.154 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 1.281 |
