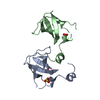
Entry Database : PDB / ID : 2qndTitle Crystal Structure of the KH1-KH2 domains from human Fragile X Mental Retardation Protein FMR1 protein Keywords / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.9 Å Authors Valverde, R. / Regan, L. Journal : Structure / Year : 2007Title : Fragile X mental retardation syndrome: structure of the KH1-KH2 domains of fragile X mental retardation protein.Authors : Valverde, R. / Pozdnyakova, I. / Kajander, T. / Venkatraman, J. / Regan, L. History Deposition Jul 18, 2007 Deposition site / Processing site Revision 1.0 Nov 6, 2007 Provider / Type Revision 1.1 Jul 13, 2011 Group / Version format complianceRevision 1.2 Aug 23, 2017 Group / Category Revision 1.3 Oct 30, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Structure summary Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature / pdbx_struct_conn_angle / struct_conn / struct_conn_type / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn_type.id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less Remark 999 Residues 331-375 have been deleted in construct; numbering pertains to full length protein. In PDB ... Residues 331-375 have been deleted in construct; numbering pertains to full length protein. In PDB file residue numbered consequetively 1 through 144. residue1=residue216 in full length protein.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj
 Assembly
Assembly




 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X6A / Wavelength: 0.9788, 0.9793, 0.9184
/ Beamline: X6A / Wavelength: 0.9788, 0.9793, 0.9184 Processing
Processing