 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2pvh: Structure-Based Design of Pyrazolo[1,5-a][1,3,5]triazine Derivati... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2pvh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure-Based Design of Pyrazolo[1,5-a][1,3,5]triazine Derivatives as Potent Inhibitors of Protein Kinase CK2 | ||||||
 Components Components | Casein kinase II subunit alpha | ||||||
 Keywords Keywords | TRANSFERASE / Structure-Based Drug Design / Enzyme-Inhibitor Complex / Casein Kinase II / Protein Kinase CK2 | ||||||
| Function / homology |  Function and homology information Function and homology informationphotoperiodism / inflorescence development / protein kinase CK2 complex / non-specific serine/threonine protein kinase / regulation of cell cycle / protein serine kinase activity / protein serine/threonine kinase activity / DNA damage response / ATP binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2.2 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2.2 Å | ||||||
 Authors Authors | Nie, Z. / Perretta, C. / Erickson, P. / Margosiak, S. / Almassy, R. / Lu, J. / Averill, A. / Yager, K.M. / Chu, S. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2007 Title: Structure-based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Authors: Nie, Z. / Perretta, C. / Erickson, P. / Margosiak, S. / Almassy, R. / Lu, J. / Averill, A. / Yager, K.M. / Chu, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2pvh.cif.gz | 86.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2pvh.ent.gz | 63.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2pvh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pv/2pvhftp://data.pdbj.org/pub/pdb/validation_reports/pv/2pvh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2pvjC  2pvkC  2pvlC  2pvmC  2pvnC  1om1S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 41450.465 Da / Num. of mol.: 1 / Mutation: V256A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P28523, non-specific serine/threonine protein kinase |
|---|---|
| #2: Chemical | ChemComp-P19 / 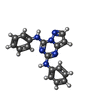  Mass: 302.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H14N6 Mass: 302.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H14N6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.79 % |
|---|---|
| Crystal grow | Temperature: 297 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 18% PEG 4000, 0.20M Sodium Acetate, 0.10M TrisHCl, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 297K |
-Data collection
| Diffraction | Mean temperature: 107 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Sep 10, 2005 / Details: MIRRORS |
| Radiation | Monochromator: OSMIC CONFOCAL MAX-FLUX MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→50 Å / Num. all: 18543 / Num. obs: 18543 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Rmerge(I) obs: 0.059 / Χ2: 0.956 / Net I/σ(I): 21.9 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.283 / Mean I/σ(I) obs: 5.1 / Num. unique all: 1796 / Χ2: 1.075 / % possible all: 97.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 1OM1 PROTEIN MODEL Resolution: 2.2→50 Å / FOM work R set: 0.847 / Isotropic thermal model: ISOTROPIC / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.24 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 35
|
