 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2oiz: Crystal Structure of the Tryptamine-Derived (Indol-3-Acetamide)-T... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2oiz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Tryptamine-Derived (Indol-3-Acetamide)-TTQ Adduct of Aromatic Amine Dehydrogenase | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / tryptophan tryptophyl quinone / H-tunneling | ||||||
| Function / homology |  Function and homology information Function and homology informationaralkylamine dehydrogenase (azurin) / aralkylamine dehydrogenase (azurin) activity / aliphatic amine dehydrogenase activity / amine metabolic process / periplasmic space Similarity search - Function | ||||||
| Biological species |  Alcaligenes faecalis (bacteria) Alcaligenes faecalis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.05 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.05 Å | ||||||
 Authors Authors | Roujeinikova, A. / Leys, D. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: New insights into the reductive half-reaction mechanism of aromatic amine dehydrogenase revealed by reaction with carbinolamine substrates. Authors: Roujeinikova, A. / Hothi, P. / Masgrau, L. / Sutcliffe, M.J. / Scrutton, N.S. / Leys, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2oiz.cif.gz | 453.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2oiz.ent.gz | 367.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2oiz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oi/2oizftp://data.pdbj.org/pub/pdb/validation_reports/oi/2oiz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2i0rC  2i0sC  2i0tC  2ojyC  2ok4C  2ok6C C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The asymmetric unit contains a biological heterotetramer |
-Components
| #1: Protein | Mass: 14516.898 Da / Num. of mol.: 2 / Fragment: (Residues: 48-182) / Source method: isolated from a natural source / Source: (natural) Alcaligenes faecalis (bacteria)References: UniProt: Q0VKG6, UniProt: P84887*PLUS, EC: 1.4.99.4 #2: Protein | Mass: 40016.125 Da / Num. of mol.: 2 / Fragment: (Residues: 73-433) / Source method: isolated from a natural source / Source: (natural) Alcaligenes faecalis (bacteria)References: UniProt: Q0VKG7, UniProt: P84888*PLUS, EC: 1.4.99.4 #3: Chemical | 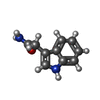  Mass: 174.199 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H10N2O Mass: 174.199 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H10N2O#4: Chemical | ChemComp-PG4 / |   Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1625 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1625 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.83 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 6 Details: PEG 2000 MME, AMMONIUM SULPHATE, SODIUM CACODYLATE, TRYPTAMINE, pH 6.0, VAPOR DIFFUSION, SITTING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Beamline: X11 |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Monochromator: TRIANGULAR HORIZONTAL- FOCUSING SI III MONOCHROMATOR Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 1.05→15 Å / Num. obs: 383635 / % possible obs: 83.6 % / Observed criterion σ(I): -3 / Redundancy: 2.6 % / Rmerge(I) obs: 0.064 / Χ2: 0.954 / Net I/σ(I): 16.5 |
| Reflection shell | Resolution: 1.05→1.07 Å / Redundancy: 2 % / Rmerge(I) obs: 0.463 / Mean I/σ(I) obs: 1.9 / Num. unique all: 16727 / Χ2: 1.056 / % possible all: 73 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.05→15 Å / Cor.coef. Fo:Fc: 0.985 / Cor.coef. Fo:Fc free: 0.98 / SU B: 0.69 / SU ML: 0.016 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.024 / ESU R Free: 0.026 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 11.547 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.05→15 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.05→1.077 Å / Total num. of bins used: 20
|
